INTRODUCTION
What better way to end the semester with a study of one of the hottest areas in signal transduction -
programmed cell death or apoptosis. We have spent time discussing how cells receive external signals from the surrounding
environment, which cause a cellular response such as differentiation, proliferation, secretion, etc. We have also seen how
important it is for cells to be able to adapt to changes and how exquisitely controlled the process of signal transduction
is. One appropriate response to a signal is for the cell to commit suicide - presumably for the good of the organism. Over
the past decade it has become very clear that programmed cell death is encoded in the genome. Apoptosis is very different
from tissue necrosis caused by an acute injury, in which cells swell and burst (from osmotic pressure differences) and cause
a significant immune response. Apoptosis consists of 4 steps:
- the decision to activate the pathway;
- the actually "suicide" of the cell;
- engulfment of the cell remains by specialized immune cells called phagocytes;
- degradation of engulfed cell.
The actual steps in cell death require:
- condensing of the cell nucleus and breaking it into pieces
- condensing and fragmenting of cytoplasm into membrane bound apoptotic bodies; and
- breaking chromosomes into fragments containing multiple number of nucleosomes (a nucleosome ladder)
To commit suicide must be an extremely important cellular decision. Hence you would expect this process
to be regulated and highly complicated. When would it be advantageous to the organism to want a cell to kill itself (or be
told to kill itself)? Cell death would be used to:
- "sculpt" an organism during development such as during embryo development, metamorphosis and tissue atrophy
- regulate the total number of cells.
- defend and remove unwanted or dangerous cells like tumor cells, virally infected cells, or immune cells
that recognize self (which coujd lead to autoimmune disease).
Unregulated apoptosis could exacerbate or cause disease such as:
- AIDS, in which T helper cell numbers plummet. Part of the dramatic decline in these cells might be caused
by health T helper cells being tricked into committing suicide;
- neurodegenerative diseases like Alzheimers;
- ischemic stroke, when restricted blood flow to certain regions of the brain can lead to neural death
through increased apoptosis'
- cancer, in which tumor cells lose their ability to undergo apoptosis;
- autoimmune disease, in which self-reactive immune cells trick normal body cells to kill themselves;
- viral disease;
Apoptosis does not require new transcription or translation, suggesting that the molecular machinery required
for cell death lay dormant in the cell, and just requires appropriate activation. What "signals" induce apoptosis?
Signals can be extracelluar:
- a hormone (such as thyroxine which causes apoptosis in tadpole tails
- a lack of a "survival" signal (which inhibits apoptosis) such as a growth factor
- a cell:cell contact from an adjacent cell
Signals can be intracelluar:
- ionizing radiation
- virus infection
- oxidative damage from free radicals
MECHANISMS AND REGULATION OF APOPTOSIS:
Caspases
Characterization of apoptotic mechanisms and cellular players started with the study of C. elegans,
a round worm. The mature worm has about 1000 cells. During development, 131 cells die. Two mutations were found in which the
131 cells did not die. These mutations were called ced3 and ced4 (ced stands for cell death). The sequence
of ced 3 was very homologous to a protein called interleukin converting enzyme (ICE) which is
required for proteolytic activation of the precursor to interleukin 1, a protein hormone released by certain immune cells
during activation and which can promote inflammation. This suggested that proteolysis was required for apoptosis. Subsequent
studies show that a whole family of proteases (about 10 in humans) called caspases (ICE has been renamed caspase 1) are required
for programmed cell death. These proteases are found in the cell in an inactive form which must undergo limited proteolysis
for activation. These caspases form a cascade of proteases which are activated in this process. They are endoproteases have
an active site Cys (C) and cleave at the C-terminal side of Asp residues (asp) and hence are known as caspases
- cys containing-asp specific proteases).
ICE is not normally involved in apoptosis, but its artificial activation in cultured mammalian cells
can lead to it. Each caspase had the same sequence as they are designed to cleave, so it became evident that they probably
cleave each other in an activation cascade mechanism, similar to the coagulation protease cascade of activation of precursors
(zymogens) of serine proteases which activate the next in the series. Two series of caspases seem to be involved. One set
initiates the process of caspase activation. Just as in the clotting system, the question of what activates the first caspase
appeared problematic until investigators found that the initiator caspase can be activated if they aggregate to a critical
concentration. This could occur by binding of a suicide signal molecule at the cell surface. Conformational changes in the
receptor can lead to aggregation of surface receptor molecules with concomitant aggregation of intracellular caspases which
interact with the aggregated receptors.
Intracellular signals
How might intracellular activators of apoptosis (like radiation or reactive oxygen species) work. Research
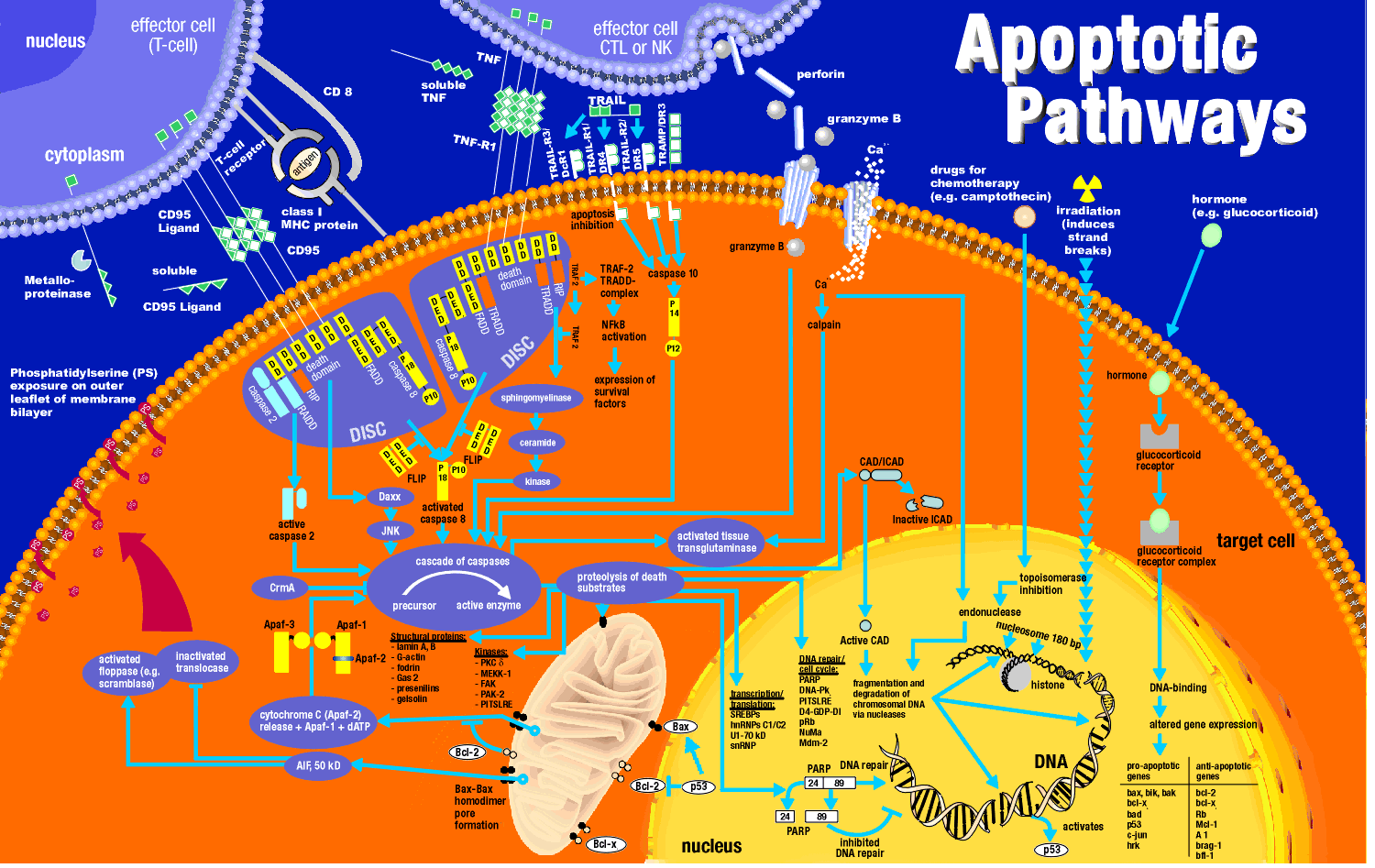
indicated involvement of mitochondria in the apoptotic pathway. Believe it or not, cytochrome C, the heme protein which
acts as a water soluble mobile carrier of electrons in mitochondrial oxidative phosphorylation, shuttling electrons through
cytochrome C oxidase or complex IV, leaks out of the intermembrane space and binds to a cytoplasmic protein called Apaf-1
for apoptotic protease activating factor-1. This then activates an initiator caspase-9
in the cytoplasm.
These proteins seem to leak out of mitochondria after a collapse of the electrochemical potential across
the inner membrane. The potential collapses as a consequence of the opening of a channel called a nonspecific inner
membrane permeability transition pore, composed of both an inner membrane protein (adenine
nucleotide translocator - ant) and an outer membrane proteins
(porin, the voltage-gated anion channel - VDAC).
These proteins act together, probably at sites where the inner and outer membranes are in contact. This channel passes
anything smaller than molecular weight 1500. Collapsing the proton gradient uncouples oxidation and phosphorylation
in the mitochondria. Changes in ionic strength causes a swelling of the matrix. Since the inner membrane is highly convoluted
and has a much greater surface area than the outer membrane, swelling of the matrix leads to a rupture of the outer membrane,
spilling the inner membrane space proteins (cytochrome C and Apaf-1) into the cytoplasm.
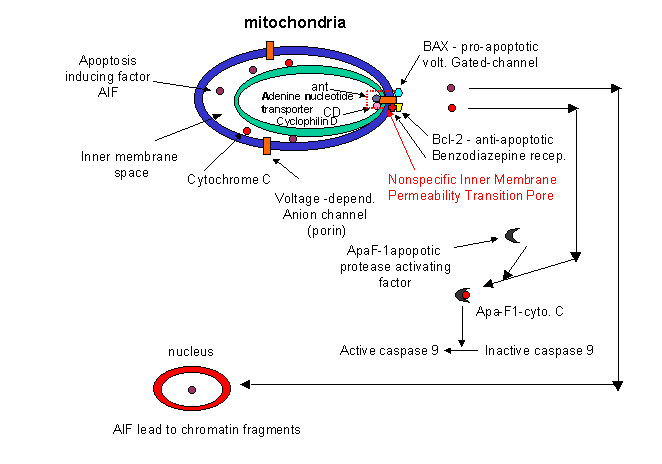
What causes all these changes in the mitochondria? Several interrelated events appear to be involved:
- disruption of ox-phos. and electron transport, caused by irradiation and certain second messengers such
as ceramide.
- changes in cell redox potential and generation of reactive oxygen species
(ROS).
- damage to DNA (caused by radiation, ROS, etc). A protein called p53 is often expressed in cells
with DNA damage. Expression of this protein results in inhibition of cell division, or apoptosis, both of which would
keep the damaged cell from becoming a tumor cell. Hence the p53 gene is a tumor suppressor gene. It is inactivated
by mutation in approximately 50% of all human tumor cells studied. p53 can induce gene expression. Of the 14 different
genes whose expression are significantly altered by p53, many seem to be used by cells to generate or respond to oxidative
stress. Cells undergo p53 apoptosis through oxidative damage.
- increases in intracellular calcium ions through signal transduction.
Caspase targets:
Apoptosis involves:
- condensing of the cell nucleus and breaking into pieces
- condensing and fragmenting of cytoplasm into membrane bound apoptotic bodies
- breaking chromosomes into fragments containing multiple number of nucleosomes (a nucleosome ladder)
How does caspase activation lead to these events? A protein has been uncovered which when cleaved by a
caspase leads to nuclear breakup. The target protein is usually bound to another protein, a DNA endonuclease. When the target
protein is cleaved, the DNase is free to migrate to the nucleus and begin the execution. Membrane changes in apoptosis occur
when caspase 3 cleaves gelsolin, a protein involved in maintaining cell morphology. The cleaved gelsolin cleaves actin filaments
inside the cell. Another protein is necessary to form apopotic bodies: a kinase named p21-activated kinase 2 (PAK-2).
This kinase is activated by caspase-3 by limited proteolysis. Caspases also cleave beta-amyloid precursor protein which
might generate more beta-amyloid protein, causing neural cell death in Alzheimer patients.
Controlling Apoptosis
It should be clear that cells keep a tight control on the caspases. Two players which appear to inhibit
apoptosis are the mitochondrial proteins Bcl-2 and Bcl-X, which can block the release of cytochrome C from the
mitochondria. The Bcl family of proteins have a hydrophobic tail and bind to the outside surface of mitochondria and other
organelles like the nucleus and endoplasmic reticulum. These proteins seem to be able to form ion channels in liposomes.
So far 15 members of this family (related to ced-9 of C. elegans) have been discovered in humans. Bcl-2 can also bind to Apaf-1
(mentioned above) and inhibit its activation of initiator caspase-9. Bcl-2 is regulated by changes in the expression of the
Bcl-2 gene, by post-translational phosphorylation by kinases, or by cleavage by caspases. Overexpression of Bcl-2 can cause
a cell to become a tumor cell. Another member of the family, BAX and BAD bind to mitochondria and facilitate
apoptosis by stimulating cytochrome C release.
In addition, other proteins called IAP's (inhibitors of apoptosis) can inhibit caspase
or other apoptotic proteins. Some virus make the protease to keep their host cells viable.
Cell Membrane Events
Cells can be instructed to undergo apoptosis through cell surface interactions with other cells which
are often immune cells. One of the jobs of the immune cell is to destroy an altered cell (for example a virally-infected
cell or a tumor cell). Immune cells themselves must also die after they are activated in an immune response. Activated
lymphocytes (like cytotoxic T cells or natural killer cells) can target and kill cells using several ways which can involved
apoptosis. In one, an activated lymphocyte binds to a target cell (like a virally infected cell) and secrete perforin,
a protein which assemble in the target cell membrane to form a transmembrane channel. Other proteins released by the
activated lymphocyte can enter the target cell through the pore and initiate apoptosis. One such protein that enters,
granzyme B is a protease which activates caspases in the target cell.
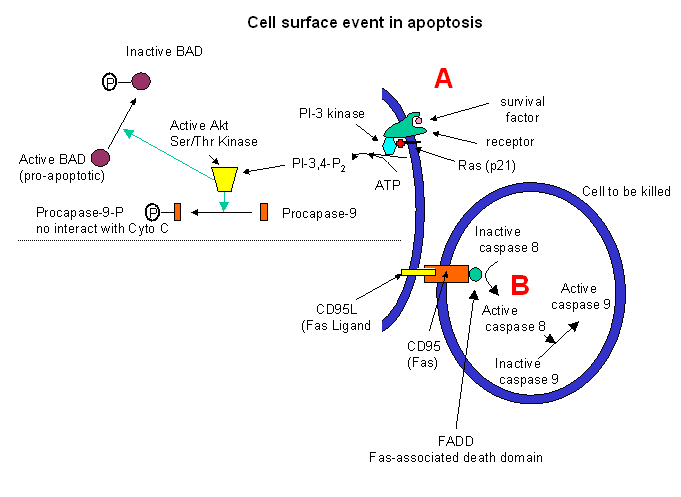
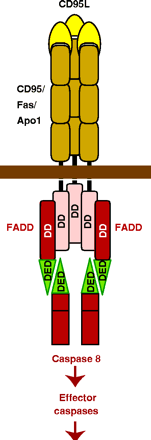
Target cells which express a specific membrane protein call CD95 (also called
Fas) are also targeted for apoptosis. This protein receptor, a member of the tumor necrosis factor receptor
(TNFR) binds to a membrane protein ligand on the surface of an activated lympocyte called CD95 Ligand - CD95L- (also
called the Fas ligand). On binding, the CD95 (Fas) receptors on the target membrane aggregate after
conformation changes. An adapter protein in the cell, FADD (Fas-associated death domain) binds to the aggregated
cytoplasmic domain (the death domain) of CD95 (Fas), and recruits inactive caspase-8 to the site, where their concentration
increases. This leads to activation of the caspases.
This mechanism is used to get rid of activated lymphocytes after they have finished their work. Activated
immune cells start expressing Fas a few days after activation, targeting them for elimination. Some cells which have
been stressed express both Fas and Fas ligand and kill themselves. Various cells express CD95 (Fas), but CD95L (Fas-Ligand)
is expressed predominately by activated T cells.
Cell surface events also can inhibit apoptosis. Binding of "survival" factors (like growth factors)
to cell surface receptors can shut of apoptotic pathways in the cells. Some survival factor receptors are coupled to
PI-3-kinase (phosphoinositol-3-kinase) through the G protein ras (p21) which is targeted
to the cell membrane by post-translational addition of a hydrophobic anchor. The activated kinase produces PI-3,4-P2
and PI-3,4,5-P3, which activates Akt, a Ser/Thr protein kinase. This activated kinase phosphorylates
the proapoptotic-protein BAD, which then becomes inactive. In addtion, active Akt phosphorylates procapse, which in
its phosphorylated form will not interact with cytochrome C, hence inhibiting apoptosis.
The endpoint of apoptosis is the engulfment of the fragmented cell by a phagocytic cell (such as a macrophage).
In a recent article (Nature, 405, pg 85, 2000), it was shown that the activity of phagocytes could be inhibited sterospecifically
by the addition of phosphatidyl serine (PS) to the mixture, but not by other negative phospolipids. If you
remember from our description of lipids, PS is found exclusively in the inner leaftlet of red blood cells). The investigators
cloned a gene from the phagocytic cell for a receptor which recognizes PS. When added to ordinary T and B lymphocytes
(immune cells), these cells could also take-up apoptotic cells. The gene is homologous to genes in Drosophila (fruit
fly) and C. elegans (round worm) suggesting that it is conserved in nature. The message: when cells undergo
apopotosis, PS, normally found only in the inner leaftlet, is exposed to the outside. It can then bind to receptors
on phagocytic cells to complete the process of apoptosis.
{kind=link}