Reactions in solution that are not catalyzed are slow since charge development and separation occurs in
the transition state. When bonds are made or broken, charged intermediates are often formed which are higher in energy than
the reactants. Since the intermediate is higher in energy than the reactants, the transition state would be even higher in
energy, and hence more closely resemble the charged intermediate. Anything that can stabilize the charges on the intermediate
and hence the developing charges in the transition states will lower the energy of the transition state and catalyze the reaction.
In this section will will investigate the mechanism underlying the catalysis by small molecules of chemical reactions.
Presumably, biological macromolecular catalyst (like protein enzymes) will use similar mechanisms in their catalytic effects
(which will be discussed in the next section).
Catalysts, including enzymes, can employ at least 5 different ways to stabilize transition states.
1. GENERAL ACID/BASE CATALYSIS
Charge development in the TS can be decreased by either donation of a proton from general acids (like
acetic acid or a protonated indole ring) to an atom such as a carbonyl O which develops a partial negative charge in the TS
when it is attached by a nucleophile. Proton donation decreases the developing negative in the TS. Alternatively, a nucleophile
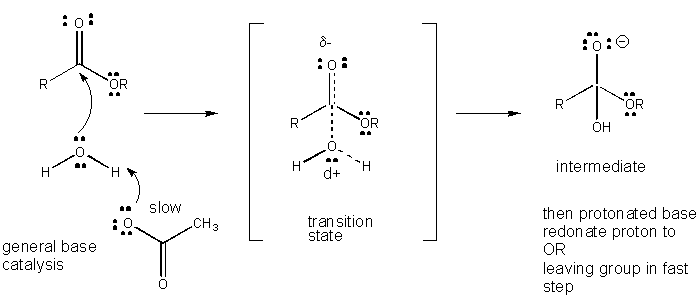
such as water which develops a partial positive charge in the TS as it begins to form a bond to an electrophilic C in a carbonyl
can be stabilized by the presence of a general base (such as acetate or the deprotonated indole ring). Proton abstraction
decreases the developing positive charge
Figure: CHARGE DEVELOPMENT IN THE TRANSITION STATE FOR ESTER HYDROLYSIS
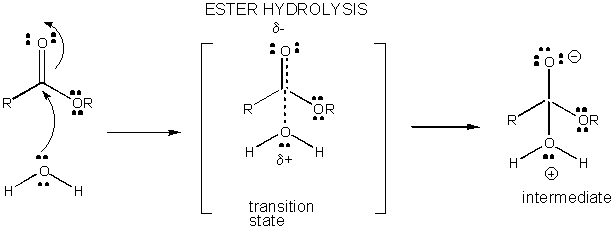
Figure: MECHANISM OF GENERAL ACID CATALYSIS
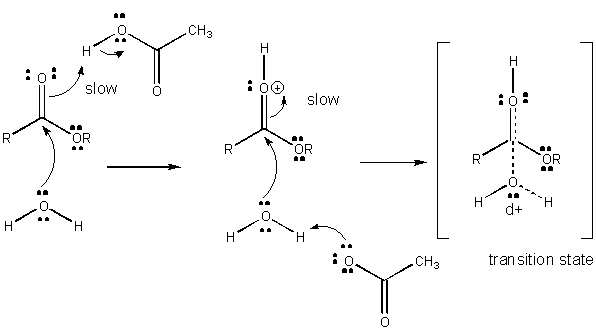
Figure: MECHANISM OF GENERAL BASE CATALYSIS
2. METAL ION (OR ELECTROSTATIC) CATALYSIS
A metal such as Cu2+ or Zn2+ can also stabilize the TS. The metal must be able
to be bound to the charged intermediate and hence the TS. The tetrahedral oxyanion intermediate of the reaction of an electrophilic
carbonyl C can interact with a metal if there is an O on an adjacent atom which can help coordinate the metal ion. This charge
stabilization of the developing negative in the TS and the full negative in the intermediate is often called electrostatic
catalysis. This method is likely to be found in many enzymes since nearly 1/3 of all enzymes require metal ions. A classic
example of an enzyme using metal ion catalysis is carboxypeptidase A.
Figure: METAL ION OR ELECTROSTATIC CATALYSIS - STABILIZATION OF TS CHARGE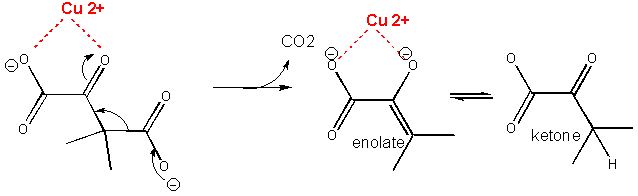
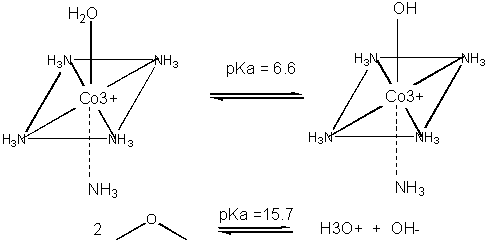
Metals can also act in a different way. They may coordinate a water and by further polarizing the H-O
bond increase the acidity of the bound water. For instance, the water molecule in the pentammineaquacobalt(III) ion has a
pKa of 6.6, compared to pure water, with a pKa of 15.7. (To calculate the latter, write the equilibrium expression for water:
H2O + H2O <==> H3O+ + OH- . Then write the Ka expression,
as for a generic acid, which is [H3O+][ OH-]/ [H2O] = 10-14/55.5. The
pKa is 15.7). The complexed hydroxide is a better nucleophile than bulk water. An example of an enzyme whose bound metal increases
the nucleophilicity of water is carbonic anhydrase.
Figure: METAL ION CATALYSIS - DECREASING pKa OF WATER
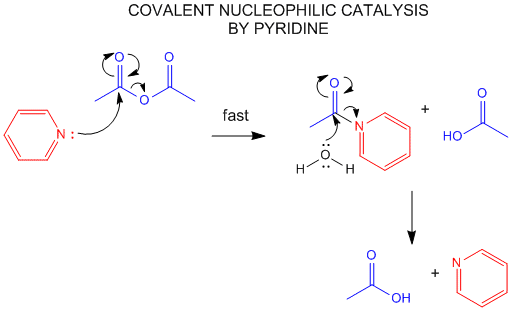
3. COVALENT OR NUCLEOPHILIC CATALYSIS
One way to change the activation energy of the reaction is to change the reaction mechanism in ways which
introduces new steps with lower activation energy. A typical way is to add a nucleophilic catalyst which forms a covalent
intermediate with the reactant. The original nucleophile can then interact with the intermediate in a nucleophilic substitution
reaction. If the nucleophilic catalyst is a better nucleophile than the original nucleophile (usually water) then the
reaction is catalyzed. The nucleophilic catalyst and the original nucleophile usually interact with a carbonyl C in a substitution
reaction, initially forming the tetrahedral oxyanion intermediate.
Figure: NUCLEOPHILIC COVALENT CATALYSIS BY PYRIDINE.
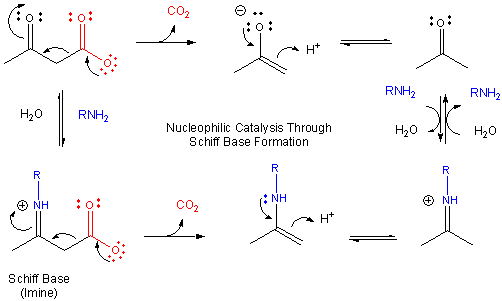
If an amine is used as the nucleophilic catalyst, then the initial addition product (a carbinolamine)
can become dehydrated, since the free pair of electrons on the N are more likely to be shared with the carbon to form a double
bond than electrons from the original carbonyl O, which is more electronegative than the N). An imine
or Schiff Base forms, with a pKa of about 7.
Figure: MECHANISM OF SCHIFF BASE FORMATION
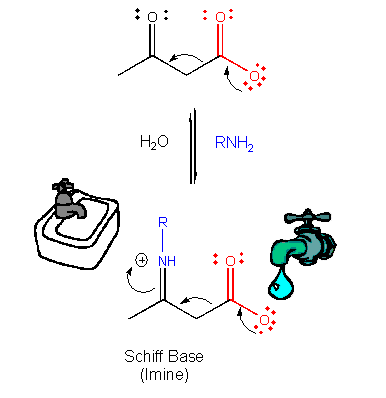
This is easily protonated to form a positively charged N at the former carbonyl O center. This serves
as an excellent electron sink for decarboxylation reactions of beta-keto acids and illustrates an important point. Electrons
in chemical reactions can be viewed as flowing from a source (such as a carboxyl group) to a sink (such as an
nucleophilic carbonyl O or a positively charged N in a Schiff base).
Figure: MECHANISM OF NUCLEOPHILIC CATALYSIS BY AMINES - SCHIFF BASE FORMATION.
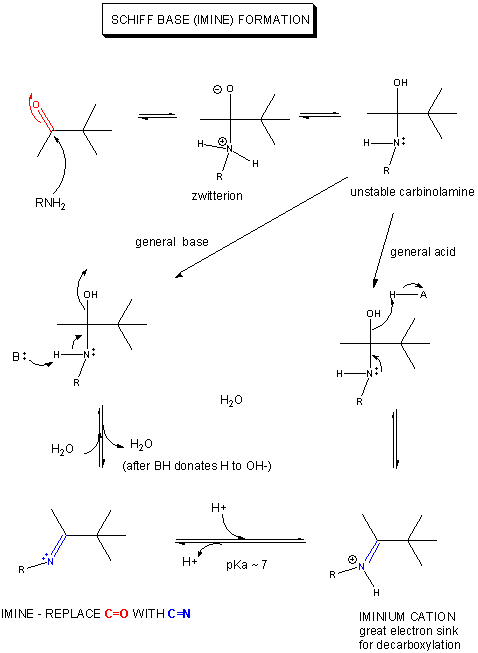
Figure: ELECTRON FLOW: SOURCE TO SINK
In a subsequent section, we will discuss how protein enzymes use these same catalytic strategies.
An intriguing question arises: how much of the structure of a large protein is really needed for catalysis? Much work
has been directed to the development of small molecule catalysis mimetics of large protein enzymes. Just how small can
you go in reducing the size of a protein and still get catalysis. One important feature of enzyme catalysis is that
they catalyze reactions in which only one enantiomer is produced. That is, the synthesis is assymertric.
This is typically a consequence of the asymmetric enzyme (itself chiral) binding only one enantiomer as a reactant and/or
the imposition of steric restrictions on the possible reactions of the bound substrate. Recently, it has been show that
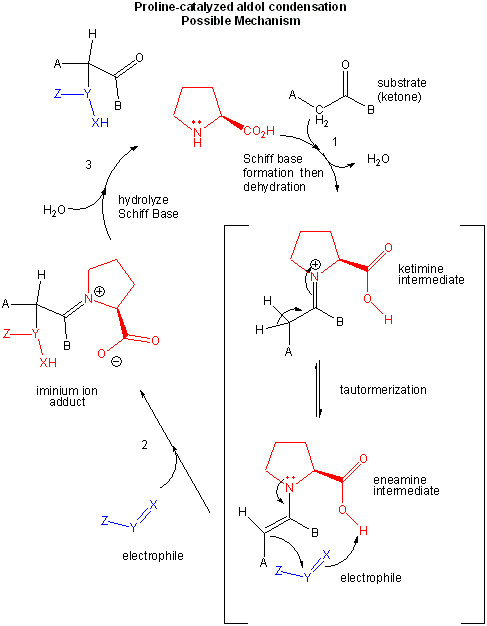
L-Pro alone can act as such an assymetric catalyst in an aldol condensation reaction.
Figure: L-PRO CATALYSIS OF AN ALDOL CONDENSATION: POSSIBLE MECHANISM
4. INTRAMOLECULAR CATALYSIS
Consider the hydrolysis of phenylacetate. This reaction, a nucleophilic subsitution reaction, could be
catalyzed by the addition to solution of the general base acetate, as described above. Since this reaction would double with
the doubling of the solution acetate, the reaction is bimolecular (first order in reactant and catalyst). Now consider the
same reaction only when the the general base part of the catalyst, the carboxyl group, is part of the reactant phenylacetate.
Such a case occurs in the acetylated form of salicylic acid - i.e. aspirin. When the carboxy group is ortho compared to the
acetylated phenolic OH, it is in perfect position to accept a proton from water, decreasing the charge development on the
O in the transition state. The general base does not have to diffuse to the appropriate site when it is intramolecular with
respect to the carbonyl C of the ester link. The rate of this intramolecular base catalysis is about 100 fold greater than
of an intermolecular base catalyst like acetate. It is as if the effective concentration of the intramolecular carboxyl base
catalyst is much higher due to its proximity to the reaction site.
Another type of reactions involving a carboxyl group (in addition to simple proton transfer) is when the
negatively charged carboxyl O acts as a nucleophile and attacks an electrophilic carbonyl carbon. When the carbonyl is part
of an ester, the carboxyl group engages in a nucleophilic substitution reaction, expelling the alcohol part of the ester as
a leaving group. The remaining examples below consider the nucleophilic (carboxyl) substitution on phenylesters, with phenolate
as the leaving group. The reactions in effect transfer an acyl group to the carboxyl group to create an anhydride.
First consider acyl transfer with aspirin derivatives. Aspirin, as you know, contains a carboxyl group
ortho to an ester substitutent. Hence the carboxyl group can act as a nucleophile and attack the carbonyl carbon of the ester
in a nucleophilic substitution reaction. The net effect is to transfer the acetyl group from the phenolic OH to the carboxyl
group converting it to an anhydride. This is an intramolecular reaction. Compare this reaction to a a comparable bimolecular
reaction shown below.
Figure: ACYL TRANSFER IN ASPIRIN DERIVATIVES.
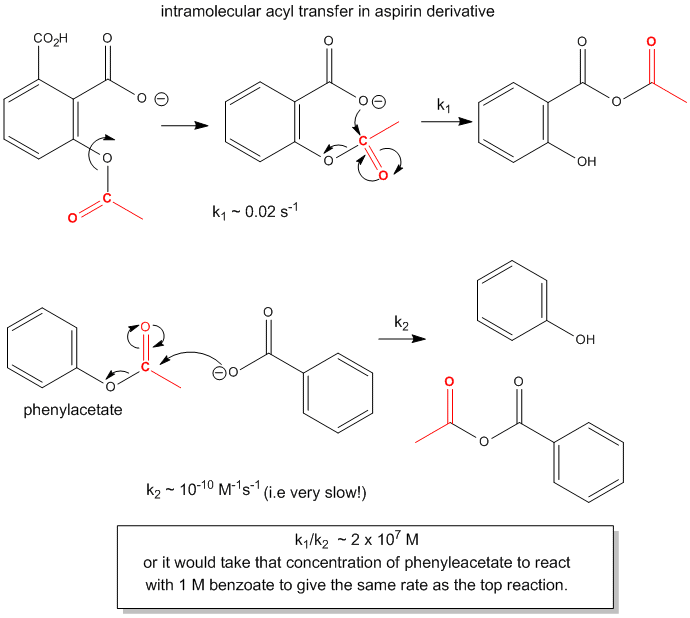
The first order rate constant of the intramolecular transfer of the acetyl group to the carboxyl group,
k1 = 0.02 s-1. The analogous bimolecular reaction rate constant k2~ 10-10 M-1s-1.
Dividing k1/k2 gives the relative rate enhancement of the intramolecular over the intermolecular
reaction. With units of molarity, this ratio can be interpreted as the relative effective concentration of the intramolecular
nucleophile. This makes the effective concentration of the carboxylate in the aspirin derivative 2 x 107
M.
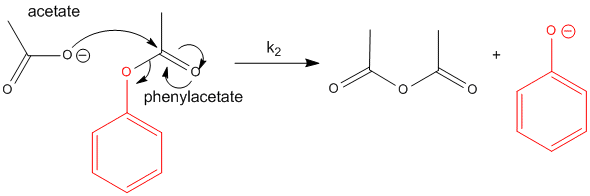
Now consider the cleavage of phenylacetate using acetate as the nucleophile. The products are acetic anhydride
and phenolate. This is a bimolecular reaction (a slow one at that), with a bimolecular rate constant, k2 which
I will arbitrarily set to 1 for comparison to some similar reactions.
Figure: REACTION OF ACETATE WITH PHENYLACETATE
Now consider a monoester derivatives of succinic acid - phenyl succinate - in which the free carboxyl
group of the ester attacks the carbonyl carbon of the ester derivative.
Figure: INTRAMOLECULAR REACTION OF PHENYLSUCCIATE
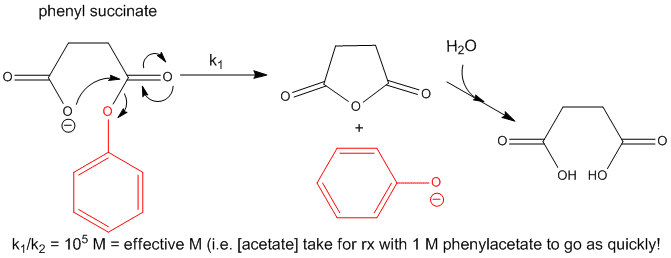
If you assign a second order rate constant k2 = 1 M-1s-1 to the analogous
intermolecular reaction of acetate with phenylacetate (as described above), the first order rate constant for the intramolecular
reaction of phenylsuccinate is 105 s-1. The ratio of rate constants, k1/k2 = 105
M. That is it would take 105 M concentration of acetate reacting with 1 M phenylacetate in the first bimolecular
reaction to get a reaction as fast as the intramolecular reaction of phenylsuccinate. An even more sterically restricated
bicyclic phenylcarboxylate shows a k1/k2 = 108 M.
Figure: INTRAMOLECULAR REACTION OF BICYCLIC PHENYLCARBOXYLATE
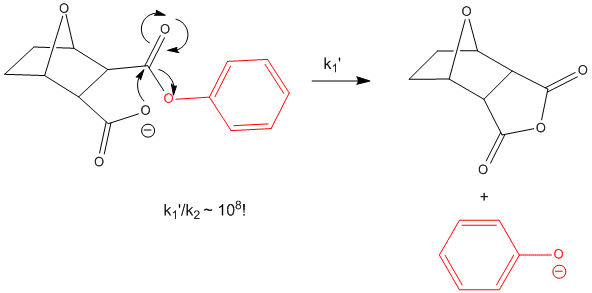
Another example is anhydride formation between two carboxyl groups. The DGo for such a reaction is positive, suggesting an unfavorable reaction. Consider two acetic acid molecules
condensing to form acetic anhydride. For this intermolecular reaction, Keq = 3x10-12 M-1.
Now consider the analogous intramolecular reaction of the dicarboxylic acid succinic acid. It condenses in an intramolecular
reaction to form succinic anhydride with a Keq = 8x10-7 (no units). The ratio Keq-intra/Keq
inter = 3 x 105 M. It is as if the effective concentration of the reacting groups. because they do not have
to diffuse together to react, is 3 x 105 M.
How does this apply to enzyme catalyzed reaction? Enzymes bind substrates in physical steps which are
typically fast. The slow step is chemical conversion of the bound substrate, which is effectively intramolecular. These three
kinds of reactions, intermolecular, intramolecular, and enzyme-catalysed can be broken down into two hypothetical steps, a
binding followed by catalysis.
Figure: three kinds of reactions, intermolecular, intramolecular, and enzyme-catalysed
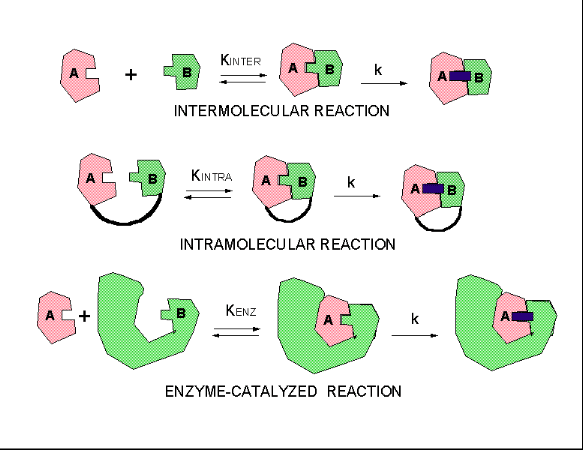
If the rate constants for the chemical steps are all identical, the advantage of the intramolecular and
enzyme-catalyzed reaction over the intermolecular reaction is KINTRA/KINTER and KENZ/KINTER,
respectively.
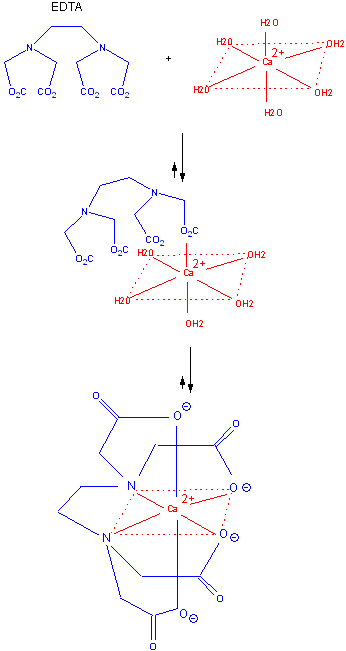
The advantage of intramolecular reactions can be seen by studying the Ca-EDTA complex. Calcium in solution
exists as a octahedrally coordinated complex with water occupying all the coordination sites. EDTA, a multidentate ligand,
first interacts through one of its potential six electron donors to Ca in a reaction which is entropically disfavored from
the the Ca-EDTA perspective, although one water is released. Once this first intramolecular complex is formed, the rest of
the ligands on the EDTA rapidly coordinate with the Ca and release bound water. The former is no longer entropically disfavored
since it is now an intramolecular process while the later is favored through the release of the remaining five water molecules.
Figure: FIGURE: BINDING OF Ca2+ AND EDTA.
We modeled the catalytic advantage offered by intramolecular reaction in terms of a dramatic increase
in the effective concentration of reactants, which sometimes reached levels of 108 M. Another way is to look
at entropy changes associated with dimer formation. The table below shows that an intramolecular reaction is favored
over an intermolecular reaction since in the latter, significant decreases in translational and rotation entropy result.
Translational, Rotational, and Internal Entropies
for Dimer Formation: A + B <=> A-B (cal/K.mol)
| System |
A |
B |
A-B |
DS |
| Gas |
| S trans |
30 |
30 |
30 |
-30 |
| S rot |
20 |
20 |
20 |
-20 |
| S int |
5 |
5 |
20 |
+10 |
| Gas -> Solution |
-10 |
-10 |
-15 |
|
| S sol |
45 |
46 |
55 |
-35 Correspond to 108-109 M) |
5. TRANSITION STATE STABILIZATION
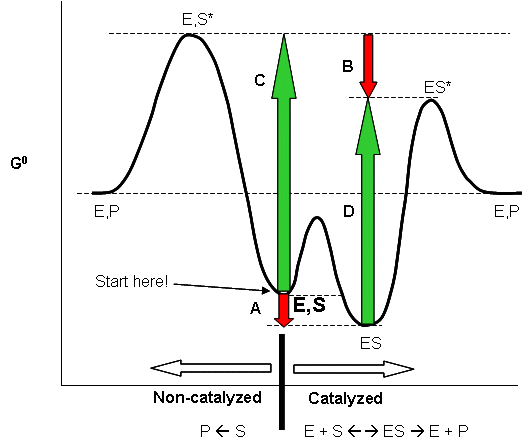
Linus Pauling postulated long ago that the only thing that a catalyst must do is bind the transition state
more tightly than the substrate. That this must be the case can be seen from the diagram below, which shows how S and S*
(the transition state) can react with E to form a complex which then proceeds to product, or can go to product in the absence
of E. From this diagram, it should be evident that c - a = d -b, where a is the DGo
for the binding of S to E, and b is the DGo for the binding of
S* to E. For an enzyme to be a catalyst the activation energy for the reaction in the presence of E, d, must be
less than in the absence of enzyme, c. Therefore c-d = a-b > 0. Since DGo
= -RTln Keq, Keq for binding of S* to E is greater than for S binding to E.
Figure: ENZYMES BIND THE TRANSITION STATE MORE TIGHTLY THAN THE SUBSTRATE
The stability of the transition state also affects the reaction kinetics (which makes sense given that the activation
energy clearly affects the speed of a reaction). As you probably remember from organic chemistry, SN2 reactions
are slow when the central atom where the substitution will occur is surrounded by bulky substitutents. (Sterics once
again.) We discussed this in context to nucleophiliic substitution on a sp2 hybridized carbonyl carbon in
carboxylic acid derivatives versus on a sp3 hybridized phosphorous in phosphoesters and diesters. The explanation
for this phenomena has usually been attributed to hindered access of the central atom caused by bulky substituents (intrinsic
effects). Is this true? Recent studies on SN2 reactions of methylchloroacetonitrile and t-butylchloroacetonitrile
(with the reagent labeled with 35Cl) using 37Cl- as the incoming nucleophile in the gas phase
shown that the more hindered t-butyl derivative's activation energy was only 1.6 kcal/mol higher than the methyl derivative,
but in aqueous solution, the difference is much greater for comparable reactions. They attributed the differences to
solvation effects of the transition state. The bulkier the substituents on the central atom, the more difficult it is
to solvate the transition state since water can't reorient around it as well. In effect there is steric hindrance for
both reactant and solvent.
ABYZMES - ANTIBODY CATALYSIS
What does it take for a macromolecule (M) to be a catalyst - an enzyme. It seems the minimum criterion
are:
- M binds a reactant
- M binds the transition state more tightly than the substrate
Anything above these is just "icing on the cake". If different functional group are present in the "active"
site of the enzyme that would allow electrostatic, intramolecular, covalent, general acid and/or base catalysis, the better
the catalyst.
Linus Pauling recognized the two key factors decades ago. He made the following hypothesis: Antibody molecules
(immune system proteins that bind foreign molecules) that can be made to bind to transition state analogs of a substrate,
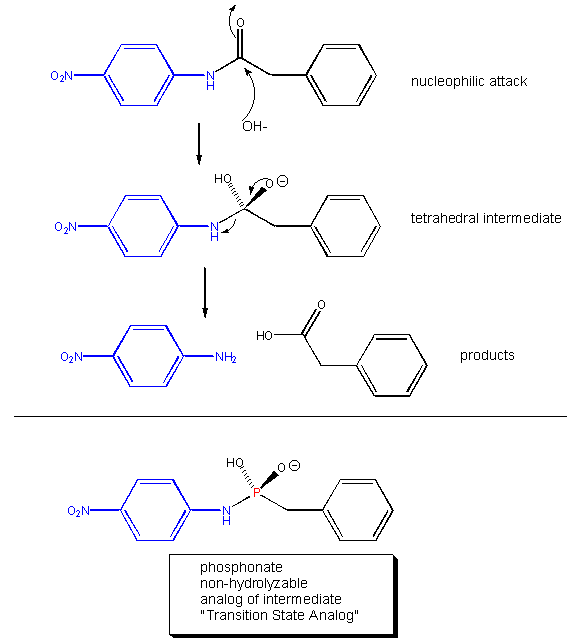
should also presumably catalyze the conversion of substrate, through the transition state, to product. About a decade ago,
his prediction was verified. Lerner et al. made a transition state analog of an ester. When an ester is hydrolyzed, the sp2
hybridized carbonyl carbon is converted to an sp3 hybridized center in the intermediate, with the carbonyl oxygen
becoming an oxyanion. The transition state presumably looks more like this unstable intermediate (sp3, oxyanion).
Lerner synthesized a phosphonate, an ester mimic with a sp3 hybridized phosphorous replacing the carbonyl C. It
also has a negatively charged oxygen as does the intermediate for the ester. This phosphonate ester is very resistant to hydrolysis.
When injected into a mouse (after first being covalently attached to a carrier protein so the small molecule becomes "immunogencic"),
the mouse makes a protein antibody which binds to the phosphonate. When the corresponding carboxylic acid ester is added to
the antibody, it is cleaved with nominal kcat and Km values. Site specific mutagenesis can then be done to make
it an even better catalyst! The antibody enzymes have been called abzymes.
Figure: PHOSPHONATES: TRANSITION STATE ANALOGS
{kind=link}