REVIEW OF ELEMENTARY REACTION KINETICS
We will briefly review various reaction orders for noncatalyzed reactions.
First Order Reaction:
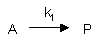
where k1 is the first order rate constant. For these reactions, the velocity of the reaction,
v, is directly proportional to [A], or
1. v = -d[A]/dt = +d[P]/dT = k1[A].
The following differential equation can be written and solved to find [A] as a function of
t.
-dA/dt = k1[A] or dA/A = -k1dt
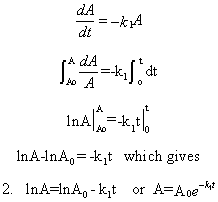
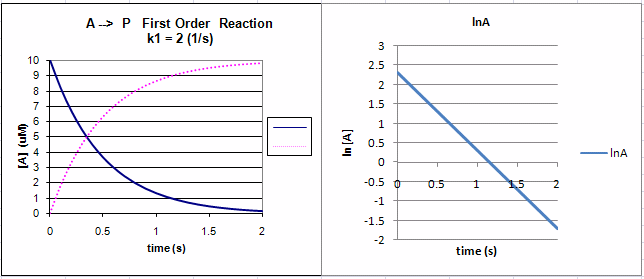
The following graphs show plots of A vs t and lnA vs t for data from a first order process.
Note that the derivative of the graph of A vs t (dA/dt) is the velocity of the reaction. The graph of lnA vs t is linear
with a slope of -k1. Note that the velocity of the reaction (slope of the A vs t curve) decreases with decreasing
A, which is consistent with equation 1.
Figure: First Order Reaction: A --> P
Second Order/Pseudo First Order Reactions
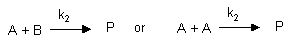
where k2 is the second order rate constant. For the first of these irreversible reactions,
the velocity of the reaction, v, is directly proportional to [A] and [B], or
3. v = -d[A]/dt = -d[B]/dt =+d[P]/dT = k2[A][B].
We will consider two special cases of this reaction type:
4. v = (k2[B]) [A] = k1' [A]
where k1' is the pseudo first order rate constant (= k2[B]
) for the reaction. The reaction appears to be first order, depending only on [A].
The following differential equation can be written and solved to find [A] as a function of
t.
5. v = -d[A]/dt = +d[P]/dT = k2[A][A] = k2[A]2
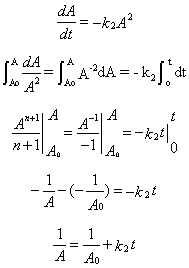
6. 1/A = 1/Ao + k2t
The following graphs show plots of A vs t and 1/A vs t for data from a second order process.
Figure: Second Order Reaction: A + A --> P
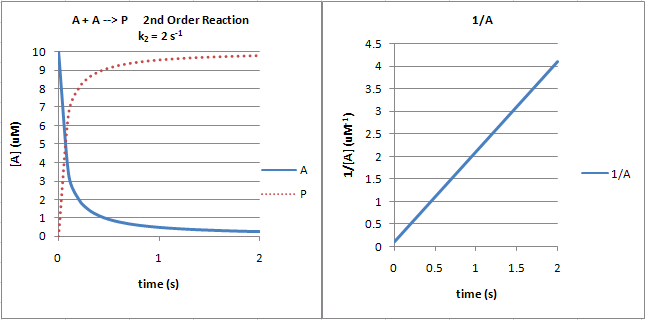
Note that just from a plot of A vs t, it would be difficult to distinguish a first from second order reaction.
If the plots were superimposed, you would observe that at the same concentration of A (10 for example as in the linked plots),
the vo of a first order reaction would be proportional to 10 but for a second order reaction to 102
or 100. Therefore, the second order reaction is faster (assuming similarity in the relative magnitude of the rate constants)
as indicated by the steeper negative slope of the curve. However, at low A (0.1 example), the vo of a first
order reaction would be proportional to 0.1 but for a second order reaction to 0.12 or 0.01. Therefore, the
second order reaction is slower.
Reversible First Order Reactions
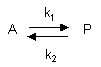
A differential equation can be written for this reaction:
7. v = -d[A]/dt = k1[A] - k2[P]
This can be solved through integration to give the following equations:
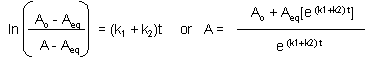
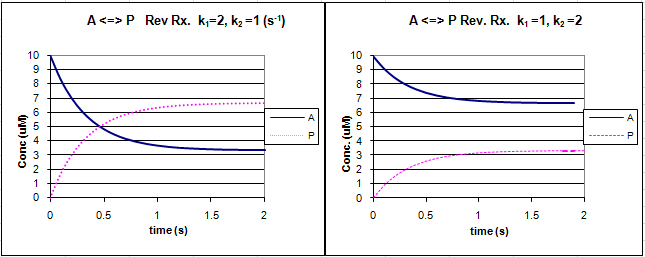
Graphs of A and P vs t for this reaction at two different sets of values of k1 and k2
are shown below.
Figure: Reversible First Order Reactions: A <=> P
Go to the following spread sheet and change the values of k1 and k2. Note the changes in the graphs.
Remember that the dissociation constant, Kd, is related to the rate constants by the formula Kd = k2/k1.
Note that if the first order rate constants are equal , Kd = Keq =1 and the equilibrium concentrations of A and P are equal.
Consecutive First Order Reactions
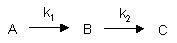
For these reactions:
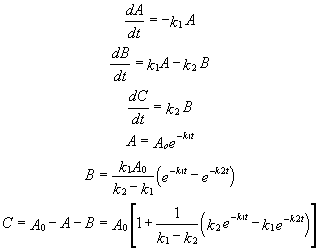
Graphs of A, B, and C vs t for these reaction at two different sets of values of k1
and k2 are shown below.
Figure: Consecutive Irreversible First Order Reactions: A --> B --> C
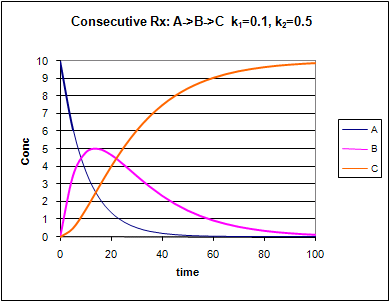
Change the values of k1 and k2. Note the changes in the graphs.
 Reactions Kinetics: Java Applet - Zero, First, and Second Order Reactions
Reactions Kinetics: Java Applet - Zero, First, and Second Order Reactions
Reaction Kinetics; Java Applet - Change forward and reverse rate constants with graphical display.
We have previously derived equations for the reversible binding of a ligand to a macromolecule. Next we
derived equations for the receptor-mediated facilitated transport of a molecule through a semipermeable membrane. This latter
case extended the former case by the addition of a physical transport step. Now, in what hopefully will seem like deja
vu, we will derive almost identical equations for the chemical transformation of a ligand, commonly referred to as a substrate,
by an enzyme. Two scenarios will be studied.
- Rapid Equilibrium Assumption - enzyme (macromolecule) and ligand (substrate) concentrations can be determined
using the dissociation constant since E, S, and ES are in rapid equilibrium, as we previously used in our derivation of the
equations for facilitated transport.
- Steady State Assumption (more general) - enzyme and substrate concentrations are not those determined
using the dissociation constant.
RAPID EQUILIBRIUM ASSUMPTION - HENRI-MICHAELIS-MENTEN EQUATION
Consider the following reaction mechanism for the enzyme-catalyzed conversion of substrate S into product
P. (We will assume that the catalyzed rate is much greater than the noncatalyzed rate.)
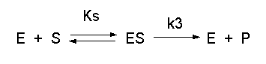
As we did for the derivation of the equations for the facilitated transport reactions under rapid equilibrium
conditions, this derivation is based on the assumption that the relative concentrations of S, E, and ES can be determined
by the dissociation constant, Ks, for the interactions and the concentrations of each species during the early part of the
reaction (i.e. under initial rate conditions). Assume also the S >> Eo. Remember that under these conditions,
S does not change much with time. Is this a valid assumption? Examine the mechanism shown above. S binds to E
with a second order rate constant k1. ES has two fates. It can dissociate with a first order rate constant
k2 to S + E, or it can be converted to product with a first order rate constant of k3 to give P + E.
If we assume that k2 >> k3 (i.e. that the complex falls apart much more quickly than S is converted
to P), then the relative ratios of S, E, and ES can be described by Ks. Alternatively, you can think about it this way. If
S binds to E, most of S will dissociate, and a small amount will be converted to P. If it does, then E is now free, and will
quickly bind S and reequilibrate since the most likely fate of bound S is to dissociate, not be converted to P (since k3
<< k2). This make sense also, if you consider that the physical step characterized by k2 is likely
to be quicker than the chemical step, characterized by k3. Hence the following assumptions have been used:
- S >> Eo
- Po = 0
- k3 is rate limiting (i.e. the slow step)
Let's assume that for this system that the initial velocity will be measured. We would like to
derive equations which show the initial velocity vo as a function of the initial substrate concentration, So
, (assuming that P is negligible over the time course of measuring the initial velocity). Also assume that the v catalyzed
>> vnoncatalyzed. In contrast to the first-order reaction of S to P in the absence of E, v is not proportional
to So but rather to Sbound. as we described in class with facilitated diffusion (flux proportional to
AR, not free A). Therefore,
vo a ES, or
Equation 1) vo = v = const [ES] = k3 [ES]
where vo is the initial velocity. How can we calculate ES when
we know S (which is equal to So) and Etot (which is Eo)? Let us assume that S is much greater
than E, as is the likely biological case. We can calculate ES using the following equations and the same procedure we used
for the derivation of the binding equation, which gives the equation below:
ES = (EoS)/(Ks + S) (analogous to ML = (MoL)/(Kd
+ L)
DERIVATION

Equation 5) v = const [ES] = k3 [ES] = k3 EoS/(Ks
+ S) = VmS/(Ks + S).
This is the world famous Henri-Michaelis-Menten Equation
It should be clear to you from this equation that:
- a plot of v vs S is hyperbolic
- v = 0 when S = 0
- v is a linear function of S when S<<Ks.
- v = Vmax (or Vm) when S is much greater than Ks
- S = Ks when v = Vmax/2.
These are the same conditions we detailed for our understanding of the binding equation
ML = (MoL)/(Kd + L)
STEADY STATE ASSUMPTION
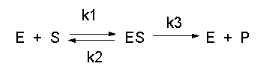
In this derivation, we will consider the following equations and all the rate constants, and will not
arbitrarily assume that k2 >> k3. We will still assume that S >> Eo and
that Po = 0. An added assumption, however, is that dES/dt is approximately 0. Look at this assumption this way.
When an excess of S is added to E, ES is formed. In the rapid equilibrium assumption, we assumed that it would fall back to
E + S faster than it would go onto product. In this case, we will assume that it might go on to product either less or more
quickly than it will fall back to E + S. In either case, a steady state concentration of ES arises within a few milliseconds,
whose concentration does not change significantly during the initial part of the reaction under which the initial rates are
measured. Therefore, dES/dt is about 0. In the rapid equilibrium derivation, we observed that v = k3ES.
We then solved for ES using Ks and mass balance of E. In the steady state assumption, the equation v = k3ES still
holds, but know we will solve for ES using the steady state assumption that dES/dt =0.
Equation 1) v = k3ES
Equation 6: (steady state) dES/dt = k1(E)(S) - k2(ES) -k3(ES)
= 0
k1(E)(S) = (k2 + k3)(ES)
k1(Eo-ES)(S) = (k2 + k3)(ES)
k1(Eo)(S) - k1(ES)(S) = (k2 + k3)(ES)
k1(Eo)(S) = (k2 + k3)(ES) + k1(ES)(S)
k1(Eo)(S) = ES (k2 + k3 + k1S)
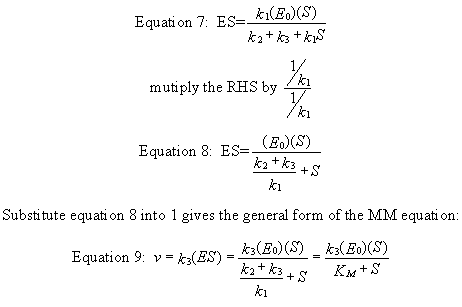
Equation 9) v = k3ES = [k3(Eo)(S)]/ [(k2
+ k3)/k1 + S] = [k3(Eo)(S)]/(Km+ S)
ANALYSIS OF THE GENERAL MICHAELIS MENTEN EQUATION
This equation can be simplified and studied under different conditions. First notice that (k2
+ k3)/k1 is a constant which is a function of relevant rate constants. This term is usually replaced
by Km which is called the Michaelis constant. Likewise, when S approaches infinity (i.e.
S >> Km, equation 5 becomes v = k3(Eo) which is also a constant, called Vm
for maximal velocity. Substituting Vm and Km into equation 5 gives the simplified equation:
Equation 10) v = Vm(S)/(Km+ S)
It is extremely important to note that Km in the general equation does not equal the Ks, the dissociation
constant used in the rapid equilibrium assumption! Km and Ks have the same units of molarity, however. A
closer examination of Km shows that under the limiting case when k2 >> k3 (the rapid equilibrium
assumption) then,
Equation 11) Km = (k2 + k3)/k1 = k2/k1
= Kd = Ks.
If we examine Equations 9 and 10 under several different scenarios, we can better understand the equation
and the kinetic parameters:
- when S = 0, v = 0.
- when S >> Km, v = Vm = k3Eo. (i.e. v is zero order with respect to S and
first order in E. Remember, k3 has units of s-1since it is a first order rate constant. k3
is often called the turnover number, because it describes how many molecules of S "turn over" to product per second.
- v = Vm/2, when S = Km.
- when S << Km, v = VmS/Km = k3EoS/Km (i.e. the reaction is bimolecular, dependent
on both on S and E. k3/Km has units of M-1s-1, the same as a second order rate constant.
Notice that equations 9 and 10 are exactly analysis to the previous equations we derived:
- ML = MoL/(Kd + L) for binding of L to M
- Jo = JmA/(Kd + A) for rapid equilibrium binding and facilitated transport of A
- vo = VmS/(Ks + S) for rapid equilbirum binding and catalytic conversion of A to P.
- vo = Vm(S)/(Km+ S) for steady state binding and catalytic conversion
of A to P.
Please notice that all these equations give hyperbolic dependencies of the y dependent variable (ML, Jo,
and vo) on the ligand, solute, or substrate concentration, respectively.
Java Applet: Michaelis-Menten Plots
EQUATION FOR MORE COMPLICATED REACTION
Not all reactions can be characterized so simply as a simple substrate interacting with an enzyme to form
an ES complex, which then turns over to form product. Sometimes, intermediates form. For example, a substrate S might interact
with E to form a complex, which then is cleaved to products P and Q. Q is released from the enzyme, but P might stay covalently
attached. This happens often in the hydrolytic cleavage of a peptide bond by a protease, when an activated nucleophile like
Ser reacts with the sessile peptide bond in a nucleophilic substitution reaction, releasing the amine end of the former peptide
bond as the leaving group, while the carboxy end of the peptide bond remains bonded to the Ser as an Ser-acyl intermediate.
Water then enters and cleaves the acyl intermediate, freeing the carboxyl end of the original peptide bond. This is shown
in the written reaction below:
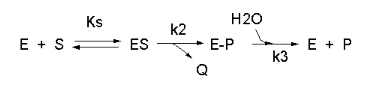
To simplify the derivation of the kinetic equation, let's assume that E, S, and ES are in rapid equilibrium
defined by the dissociation constant, Ks. Assume Q has a visible absorbance, so it is easy to monitor. Assume from the
steady state assumption that:
d[E-P]/dt = k2[ES] - k3[E-P] = 0 (assuming k3 is a
pseudo first order rate constant and [H2O] doesn't change.
The velocity depends on which step is rate limiting. If k3<<k2, then
the k3 step is rate-limiting. Then
v = k3[E-P]
If k2<<k3, then the k2 step is rate-limiting. Then
v = k2[ES]
For a homework assignment, you derived the following kinetic equation for this reaction, assuming
v = k2[ES].
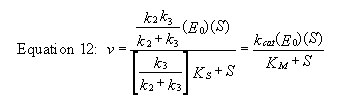
You can verify that you get the same equation if you assume that v = k3[E-P].
(Also, kcat will be defined below.)
This equation looks quite complicated, especially if you substitute for Ks, k-1/k1.
All the kinetic constants can be expressed as functions of the individual rate constants. However this equation can be simplified
by realizing the following:
- When S >> [Ks(k3)/(k2+k3)], v = [(k2k3)/(k2
+ k3)]Eo = Vm
- [Ks(k3)/(k2+k3)] = constant = Km
Substituting these into equation 7 gives:
Equation 13 - v = Vm(S)/(Km+ S)
This again is the general form of the Michaelis-Menten equation
Note in the first bulleted item above that for this reaction, Vm = [(k2k3)/(k2
+ k3)]Eo
This is more complicated than our earlier definition of Vm = k3Eo. They are similar
in that the term Eo is multiplied by a constant which is itself a function of rate constant(s). The rate constants
are generally lumped together into a generic constant called kcat. For the simple reaction kcat =
k3 but for the more complicated reaction with a covalent intermdiate which we just deived, kcat
= (k2k3)/(k2 + k3). For all reactions,
Equation 14 - Vm = kcatEo.
Figure: Summary of vo vs S equations - Meanings of Km, kcat and Vm.
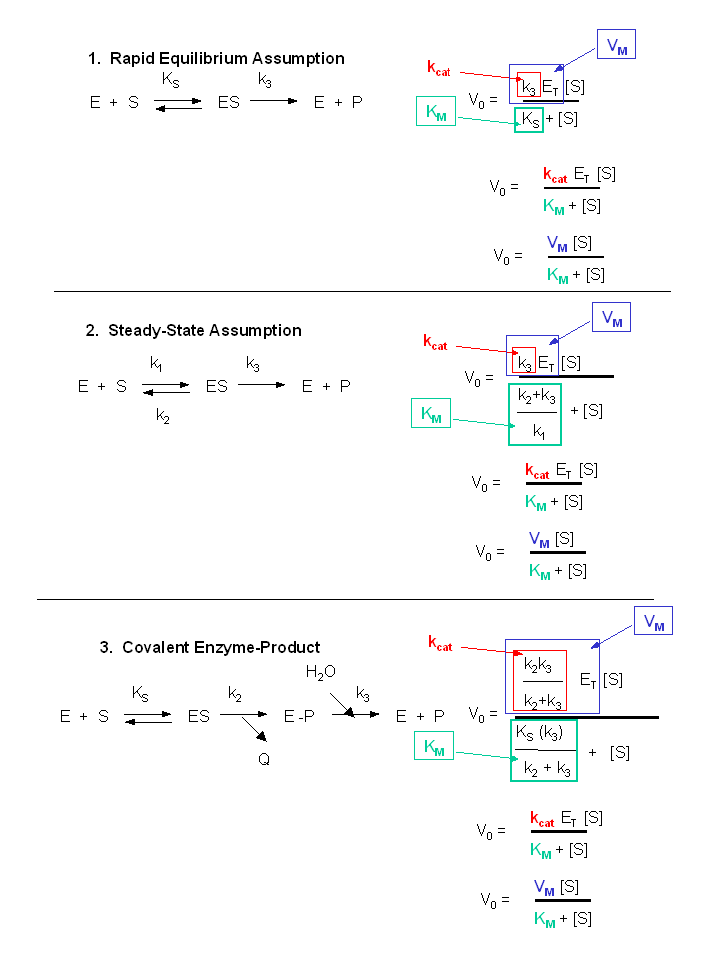
eXPERIMENTAL DETERMINATION OF Vm AND
Km
How does one extract Vm and Km
from experimental data? We have already answered this question when we explored different mathematical transformations of the binding equation to determine Kd. These included:
-
nonlinear hyperbolic fit
-
double reciprocal plot
-
Scatchard plot
The double-reciprocal plot is commonly used to analyze initial velocity vs substrate concentration data.
When used for such purposes, the graphs are referred to as Lineweaver-Burk plots, where plots of 1/v vs 1/S are straight lines
with slope m = KM/Vmax, and y intercept b = 1/Vmax. These plots can not be analyzed
using linear regression, however, since that method assumes constant error in the y axis (in this case 1/v) data. A
weighted linear regression or even better, a nonlinear fit to a hyperbolic equation should be used. The Mathcad template
below shows such a nonlinear fit. In the laboratory, we will use a series of programs developed by W. W. Cleland specifically
designed to analyze initial rate data of enzyme catalyzed reactions.
 Mathcad 8 - Nonlinear Hyperbolic Fit. Vm and Km.
Mathcad 8 - Nonlinear Hyperbolic Fit. Vm and Km.
{kind=link}