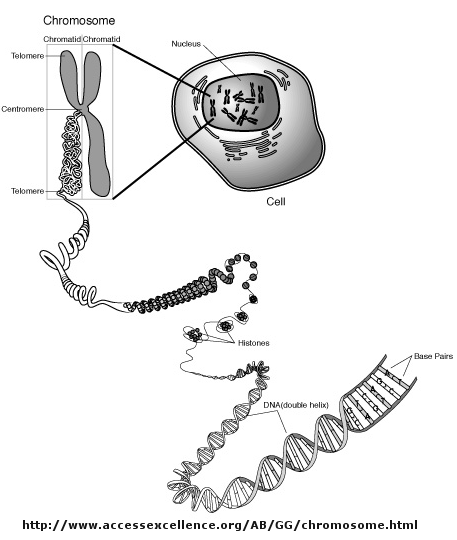
Imagine you have been given a string approximately 3 feet long, which represents an unwound, deproteinated,
human chromosome. In actuality, DNA in the nucleus is wound around a series of positively charged histone proteins; in the
electron microscope it resembles beads on a string. This structure is wound into a cylindrical "solenoid" structure which
is further packaged to fit into the nucleus along with the rest of the chromosomes. There just happens to be a small dot in
the dead center of the string you have been given. It represents the gene for a particular protein called metallothionein.
This gene is expressed and the protein metallothionein is made when cells are exposed to heavy metals like Cd. Your job today
is to figure out a possible way (there are many) in which the cell would not express the gene, or express it to a small, constituitive
level, in the absence of Cd exposure, and how the gene might be activated - induced to transcribe the gene and translated
the resulting mRNA to form the metallothionein protein upon exposure to Cd. (Hint: Cd doe not DIRECTLY bind to DNA.) Propose
mechanisms to address the following questions. Do not propose any magical mechanisms:
- How might gene expression be repressed in the absence of Cd. List some possibilities.
- How might gene expression be activated in the presence of Cd
One of the central questions of modern biology is what controls gene expression. As we have previously
described, genes must be "turned on" at the right time, in the right cell. To a first approximation, all the cells in an organism
contains the same DNA (with the exception of germ cells and immune cells). Cell type is determined by what genes are expressed
at a given time. Likewise, cell can change (differentiate) into different types of cells by altering the expression
of genes. The central dogma of biology describes how genes are first transcribed to RNA, and then the mRNA is translated into
a corresponding protein sequence. Proteins can then be post-translationally modified, localized to certain locales within
the cells, and ultimately degraded. If functional proteins are considered the end-product of gene expression, the control
of gene expression could theoretically occur at any of these steps in the process.
Figure: PROCESSES THAT AFFECT THE STEADY STATE CONCENTRATION OF A PROTEIN
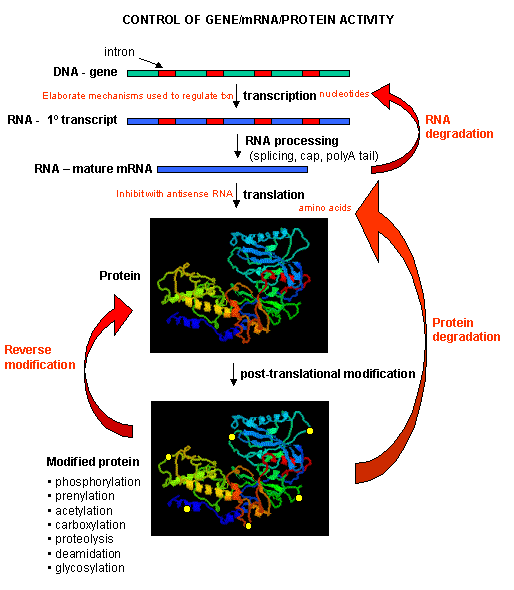
Mostly, however, gene expression is controlled at the level of transcription. This makes great biological
sense, since it would be less energetically wasteful to induce or inhibit the ultimate expression of a functional protein
at a step early in the process. How can gene expression be regulated at the transcriptional level? Many examples have been
documented. The main control is typically exerted at the level of RNA polymerase binding just upstream (5') of a a
site for transcriptional initiation. Other factors, called transcription factors (which are usually proteins), bind to the
same region and promote the binding of RNA polymerase at its binding site, called the promoter. Proteins can also bind to
sites on DNA (operator in prokaryotes) and inhibit the assembly of the transcription complex and hence transcription. Regulation
of gene transcription then becomes a matter of binding the appropriate transcription factors and RNA polymerase to
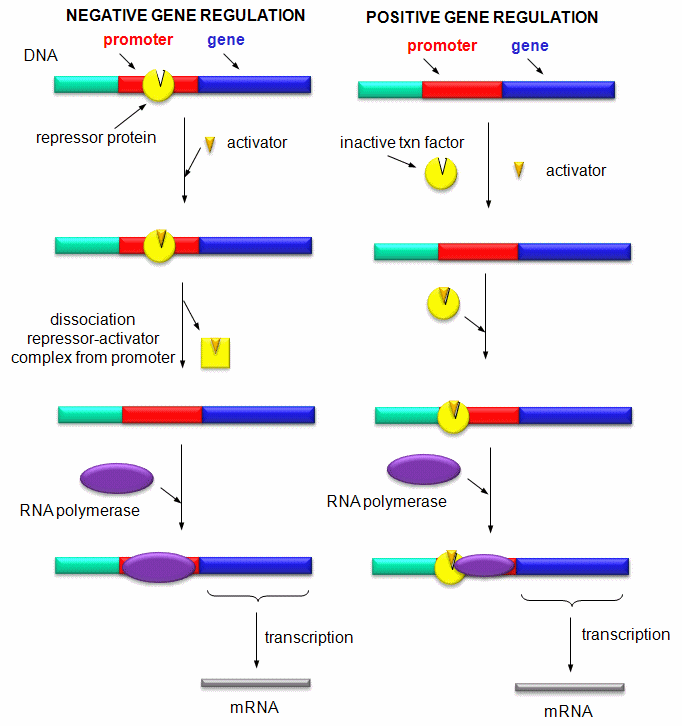
the appropriate region at the start site for gene transcription. Regulation of gene expression by proteins can be either positive
or negative. Regulation in prokaryotes is usually negative while it is positive in eukaryotes.
Figure: Positive and Negative Regulation of Gene Transcription
CONTROL OF GENE TRANSCRIPTION IN PROKARYOTES: THE E. COLI LAC OPERON
The regulation of the genes involved in lactose utilization won Jacob and Monod (of MWC fame) the Nobel
Prize. Lactose can be used as the sole source of carbon by E. Coli. Three genes are required for lactose utilization, beta-galactosidase
(lac Z, cleaves lactose to gal and glu), galactoside permease (lac Y, transports lac into the cell) and thiogalactoside transacetylase
(lac A, function unknown). These genes follow one another on the DNA, and have 1 promoter region. On transcription and translation,
one long poly-protein is made, which is cleaved post-translationally to form the individual proteins.
Figure: FUNCTION OF PROTEINS IN GALACTOSE UTILIZATION
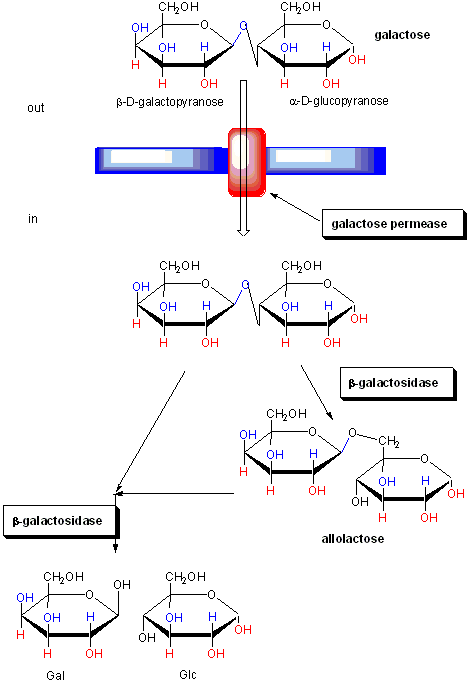
In addition, another gene, the gal repressor, is found just upstream of the gal utilization genes. It
has its own promoter (PI) The gene cluster, including promoter and any regulatory DNA sequences is called an operon,
for example, the Lac operon. In this case, the operon is induced in response to a molecular signal - i.e. the presence of
lactose, or allolactose. This binds to the repressor protein, which is bound to the operator DNA, inhibiting transcription.
When allolactose or another beta-galactosides, such as isopropylthiogalactoside (IPTG), bind to the repressor protein, a conformation
change occurs in the repressor, resulting in a higher Kd for the operator DNA, and subsequent dissociation of the repressor-galactoside
complex. Transcription ensues.
Figure: IPTG and Lactose Structures
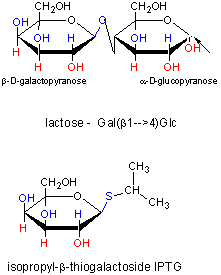
IPTG is an inducer of the lac operon but is not a substrate for the enzymes produced. We will use IPTG
to induce expression of human adipocyte acid phosphatase b (HAAP-b) in lab 4B. The plasmid containing the gene for HAAP-b has been engineered to contain the lac promoter just before the start site for gene transcription. By adding IPTG
to the growing cells, the cells can be induced to synthesize the protein, HAAP-b.
Figure: INDUCTION OF LAC OPERON
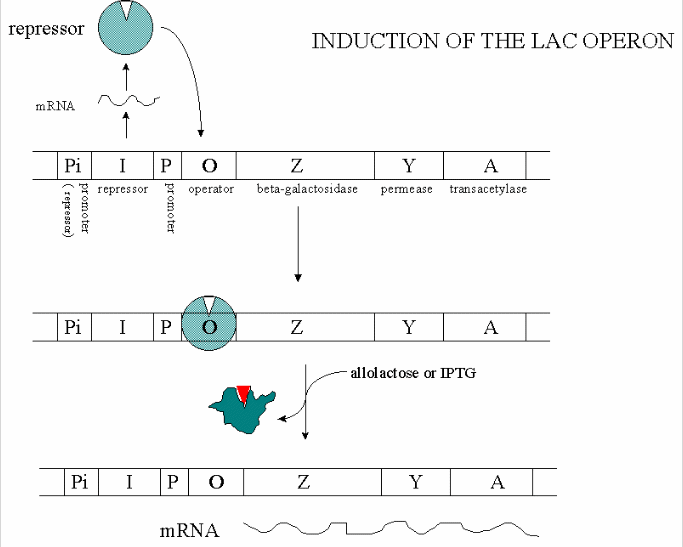
Many analogous but distinct methods are used to control gene transcription in prokaryotes. The control
of lac operon transcription is but one example.
CONTROL OF GENE TRANSCRIPTION IN EUKARYOTES
Three major differences exists in the control mechanisms used to regulate gene transcription
in eukaryotes compared to prokaryotes.
- multiple changes occur in the structure of chromatin at the site of transcription
- positive mechanisms regulate transcriptions much more often than negative ones.
- transcription and translation occur at spatially and temporally distinct sites and
times.
The genomes of eukaryotes are much larger than prokaryotes. This poses some problems with
respect to binding. Remember, DNA binding proteins demonstrate both nonspecific and specific binding. Nonspecific binding
may help a protein find a specific site in the genome, but as the size of the genome increases, the chance of finding multiple
specific sites randomly distributed increases. This problem can be avoided if multiple proteins are required to generate an
active transcription complex. The chance of finding two or more specific sites for different proteins in proximity at sites
other than required for gene transcription are very low. Multiple negative regulators would not be needed since just the binding
of one regulator would probably be sufficient. Most eukaryotic genes have about 5 regulatory sites for binding transcription
factors and RNA polymerase. Examples of these transcription factors are show in the figure below.
Figure: Control of globin gene transcription
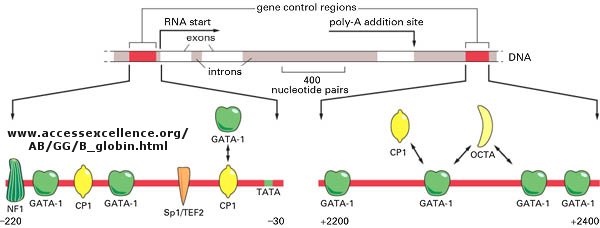
Figure: Example of transcription complexes
Light can even regulate gene expression. A great new example by Quail shows how light can change
and activate an inactive transcription factor in plants. "The basic helix-loop-helix transcription factor PIF3 binds
to a G-box motif in the promoter region of light-responsive genes. Upon absorbing red light, a phytochrome photoreceptor is
converted from the inactive Pr form to the active Pfr form, which moves to the nucleus.
Figure: Upon absorbing red light, a phytochrome photoreceptor is converted from the inactive Pr form to the active
Pfr form
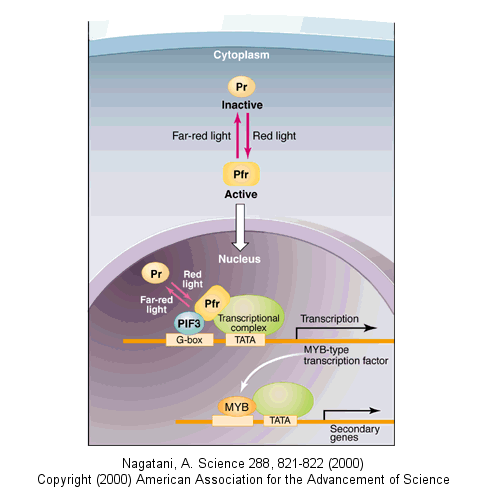
Here, Pfr is recruited to the promoter region of target genes by binding to PIF3 and then activates the
expression of genes encoding MYB class transcription factors (CCA1, LHY). The transcription factors in turn activate the expression
of secondary genes. Far-red light shuts down this signaling pathway by converting Pfr back to Pr, promoting its release from
the PIF3 complex."
STRUCTURAL FEATURES OF SPECIFIC DNA BINDING SITES
Since RNA polymerase must interact at the promoter site of all genes, you might expect that all genes
would have a similar nucleotide sequence in the promoter region. This is found to be true for both prokaryotic and eukaryotic
genes. You would expect, however, that all transcription factors would not have identical DNA binding sequences.
Prokaryotic Promoter Sequences
|
Promoter |
-35 Region |
Spacer |
-10 Region |
Spacer |
RNA start |
| trp operon |
TTGACA |
N17 |
TTAACT |
N7 |
A |
| tRNAtyr |
TTACA |
N16 |
TATGAT |
N7 |
A |
| lP2 |
TTGACA |
N17 |
GATACT |
N6 |
G |
| lac operon |
TTTACA |
N17 |
TATGTT |
N6 |
A |
| rec A |
TTGATA |
N16 |
TATAAT |
N7 |
A |
| lex A |
TTCCAA |
N17 |
TATACT |
N6 |
A |
| T7A3 |
TTGACA |
N17 |
TACGAT |
N7 |
A |
| consensus |
TTGACA |
|
TATAAT |
|
|
Eukaryotic Response Elements (RE)s
|
Regulatory agent |
Module |
Consensus |
DNA bound |
Factor |
Size (daltons) |
| Heat Shock |
HSE |
CNNGAANNTCCNNG |
27 bp |
HSTF |
93,000 |
| Glucocorticoid |
GRE |
TGGTACAAATGTTCT |
20 bp |
Receptor |
94,000 |
| Cadmium |
MRE |
CGNCCCGGNCNC |
. |
? |
. |
| Phorbol Ester |
TRE |
TGACTCA |
22 bp |
AP1 |
39,000 |
| Serum |
SRE |
CCATATTAGG |
20 bp |
SRF |
52,000 |
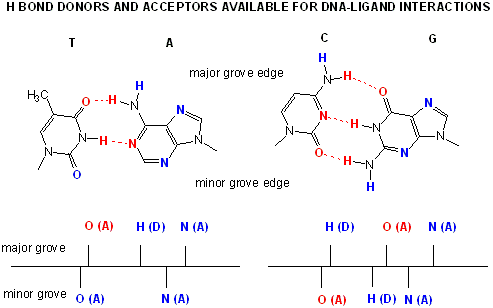
Proteins can interact specifically with DNA through electrostatic, H-bond, and hydrophobic interactions.
AT and GC base pairs have available H bond donors and acceptors which are exposed in the major and minor grove of the ds DNA
helix, allowing specific protein-DNA interactions.
Figure: AT and GC base pairs have available H bond donors and acceptors
Gene Transcription and Control of Membrane Lipids
An interesting example of transcriptional control occurs to maintain the balance of lipids in biological
membranes. The phospholipids and spingolipids in membranes are extremely heterogeneous, owing to the diversity of head
groups and acyl chain composition. Given this great diversity, it is remarkable the different cells are able to maintain
the specificity of lipid types in different cells, in different membranes within cells, and within a given leaflet of a membrane
(remember our discussion of lipid rafts). How can the cell regulate the type of lipids that it synthesizes? What
controls the transcription of genes for lipid synthesis?
Regulation of transcription of these genes appears to be controlled by proteins that bind to sterol response
elements in the DNA. The proteins, called sterol response element binding proteins (SREBPs) are activated to become
transcription factors and migrate to the nucleus when they are released from Golgi membrane by proteolysis by Golgi
proteases. The SREBP in the Golgi is in complex with another protein, SCAP, which facilitates movement of the
SREBP to the Golgi from its site of synthesis in the endoplasmic reticulum. Lipid regulation occurs when fatty acids,
cholesterol, or PL derivatives like phosphoethanolamine (from ceramide) inhibits proteolytic activation of the SREBP.
Regulation depends on whether or not SCAP "ferries" SREBP to the Golgi.
STRUCTURAL FEATURES OF DNA-BINDING PROTEINS
Not any protein can bind specifically to DNA. Analysis of DNA binding proteins shows common motifs are
found among them.
- helix-turn-helix: found in prokaryotic DNA binding proteins.
Figure: helix-turn-helix
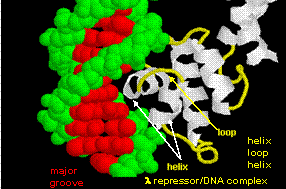
The figures shows two such proteins, the cro repressor form bacteriophage 434 and the lambda repressor
from the bacteriophage lambda. (Bacteriophages are viruses that infect bacteia.) Notice how specificity is achieved,
in part, by the formation of specific H-bonds between the protein and the major grove of the operator DNA.
Figure: Lambda Repressor/DNA Complex
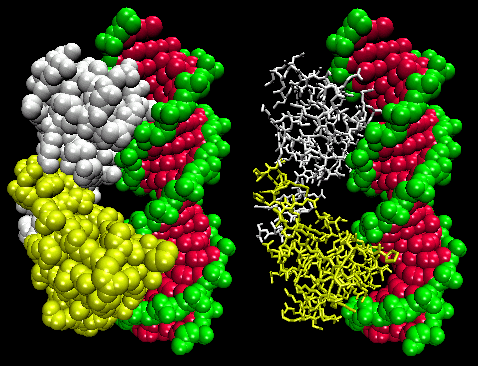
Figure: H Bond interactions between l repressor and DNA
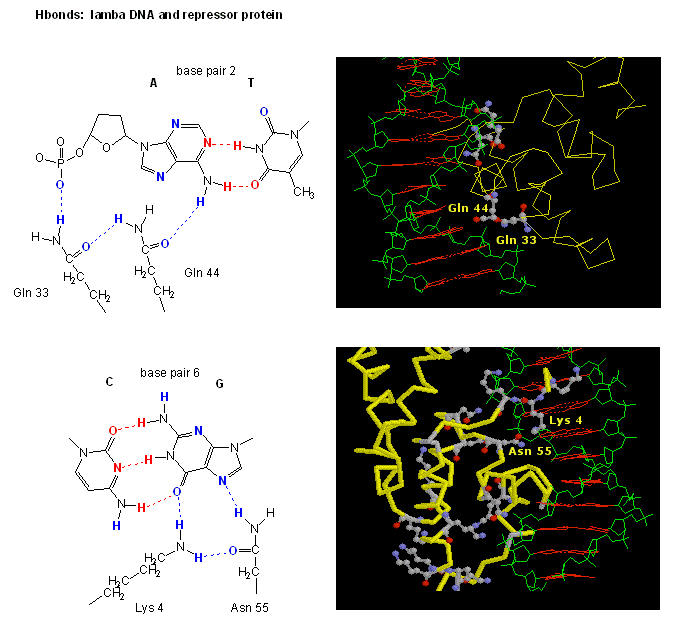
.
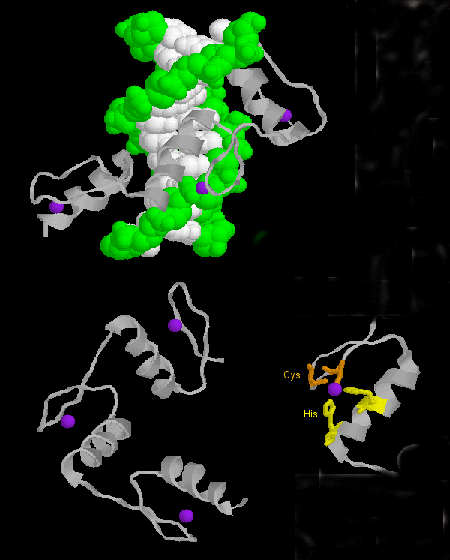
- zinc finger: (eukaryotes) These proteins have a common sequence motif of
X3-Cys-X2-4-Cys-X12-His-X3-4-His-X4-
in which X is any amino acid. Zn2+ is tetrahedrally coordinated with the Cys and His side chains, which are on
one of two antiparallel beta strands, and an alpha helix, respectively. The zinc finger, stabilized by the zinc, binds to
the major groove of DNA. ]
Figure: zinc finger
- steroid hormone receptors: (eukaryotes) In contrast to most hormones, which bind to cell surface
receptors, steroid hormones (derivatives of cholesterol) pass through the cell membrane and bind to cytoplasmic receptors
through a hormone binding domain. This changes the shape of the receptor which then binds to a specific site on the DNA (hormone
response element) though a DNA binding domain. In a structure analogous to the zinc finger, Zn 2+ is tetrahedrally
coordinated to 4 Cys, in a globular-like structure which binds as a dimer to two identical, but reversed sequences of DNA
(palindrome) within the major grove. (An example of a palindrome: Able was I ere I saw Elba.)
- leucine zippers: (eukaryotes) These proteins contain stretches of 35 amino acids in which Leu
is found repeatedly at intervals of 7 amino acids. These regions of the protein form amphiphilic helices, with Leu on one
face. Two of these proteins can form a dimer, stabilized by the binding of these amphiphilic helices to one another, forming
a coiled-coil, much as in the muscle protein myosin. Hence the leucine zipper represents the protein binding domain of the
protein. The DNA binding domain is found in the first 30 N-terminal amino acids, which are basic and form an alpha helix when
the protein binds to DNA. The leucine zipper then functions to bring two DNA binding proteins together, allowing the N-terminal
bases helices to interact with the major grove of DNA in a base-specific fashion.
Figure: leucine zippers
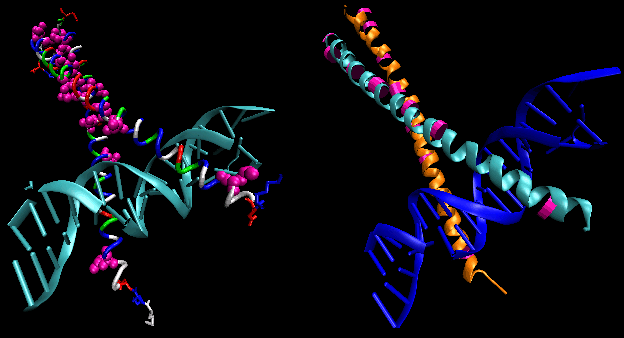
PHOSPHORYLATION AND CONTROL OF GENE EXPRESSION
A common way to control gene expression is by controlling the post-translational phosphorylation of transcription
factors by ATP. This modification might activate or inhibit the transcription factor in turning on gene expression.
The added phosphate groups might be necessary for direct binding interactions leading to gene transcription or they might
lead to a conformational change in the transcription factor, which could activate or inhibit gene transcription. A recent
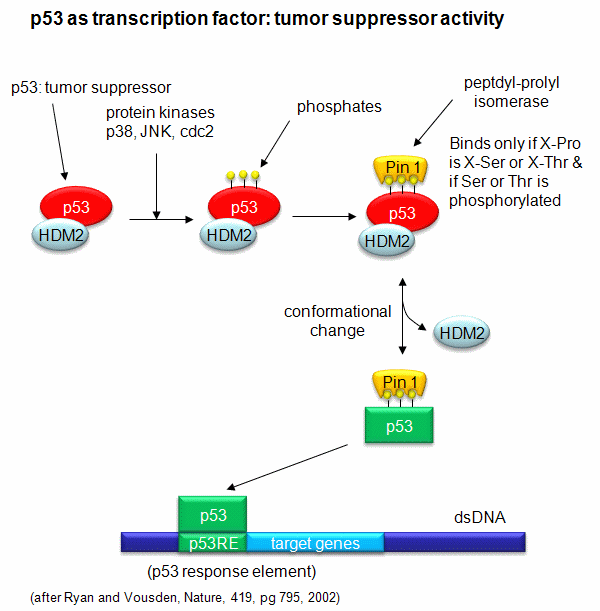
example of this later case is the control of the activity of the transcription factor p53. p53 has many activities in
the cell, a primary one as a suppressor of tumor cell growth. If a cell is subjected to stress that results in genetic
damage (an evident which could lead a cell to transform into a tumor cell), this protein becomes an active transcription factor,
leading to the expression of many genes, including those involved in programmed cell death and cell cycle regulation.
Both of these effects could clearly inhibit cell proliferation. Hence p53 is a tumor suppressor gene. p53 is usually
bound to the protein HDM2 which down regulates its activity by leading to its degradation. Stress signals lead to the
activation of protein kinases in the cell (such as p38, JNK, and cdc2), causing phosphorylation of Ser 33 and 315 and Thr
81 in p53. This leads to the binding of Pin 1, a peptidyl-prolyl isomerase, which catalyzes the trans<=>cis conformational changes of X-Pro bounds. Pin 1 appears to bind only when p53
is phosphorylated. The ensuing change in p53 conformation presumably leads to its activation as a transcription factor.
Figure: Activation of p53 as a transcription factor by phosphorylation and conformational change
POSITIVE TRANSCRIPTION FACTORS - A NEW CLASIFICATION
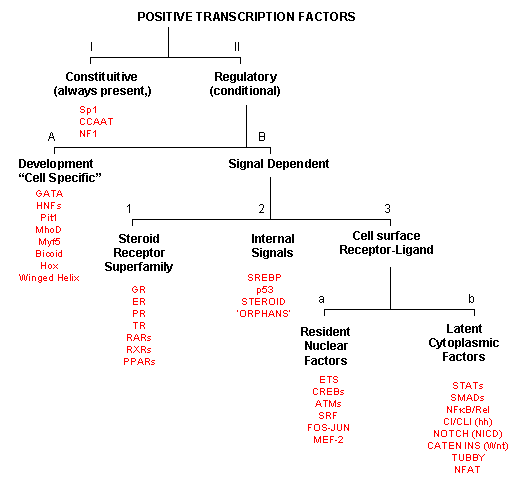
As inferred from above, transcription factors can be classified based on their protein structure.
A newer classification scheme, based on the function/activity of the transcription factors, has been proposed by Brivan, Lou
and Darnell (Science, 295, pg 813, 2002), as illustrated in the flowchart below, along with specific examples. The classes
of transcription factors include those that are:
The rest must be activated by some means, which include those that are:
-
developmental or cell type-specific whose genes must be transcribed (probably in a regulated
fashion) to form the transcription factor which then enters the nucleus;
-
signal dependent transcription factors, which are activated through a signaling event.
There are classes of signal-dependent transcription factors that are activated by:
-
steroids, which are cholesterol derivatives that can pass through the cell membrane
and bind steroid-specific transcription factors which turn on specific sets of genes; most of these transcription factors
are present in the nucleus and are activated there by steroid hormones. One exception is the glucocorticoid receptor
(GR) which is found in the cytoplasm;
-
internal signals derived from the cell, such as internally made lipid signals.
-
cell surface receptor-ligand interactions;
There are two types of receptor-ligand interactions that lead to transcription factor initiation.
-
small ligand molecules (like epinephrine) bind transmembrane receptors leads to formation of
second messengers or signals inside the cell, which ultimately activate Ser-phosphorylation activity. Nuclear transcription
factors can become phosphorylated and activated.
-
small ligands bind transmembrane receptors which then bind to and activate latent transcription
factors in the cytoplasm, which then migrate to the nucleus.
Figure: Transcription Factors: Functional Classification
COOPERATIVE BINDING OF PROTEINS TO DNA
We have just spend much time studying the cooperative binding of oxygen to hemoglobin. Cooperativity
seemed to be require conformational changes in a multimeric protein. Is it possible to get cooperative binding
of ligands without conformational changes? In a recent book by Ptashne and Gann (Genes and Signals, Cold Spring Harbor
Press, 2002), it is argued that you can and through a very simple mechanism.
It must be clear that to activate gene transcription, several transcription factor proteins must assembly
at the promoter before RNA polymerase can transcribe a gene. There are multiple DNA-protein and protein-protein contacts.
To simplify this discussion, consider the case of two proteins, A and B, that must bind to the DNA and to each other for transcription
to occur.
Figure: two proteins, A and B

The binding of each protein alone is characterized by a characteristic Kd, kon, and koff.
What happens to kon and koff for protein B, for example, when A is already bound? You can imagine
that kon doesn't change much, but what about koff after the protein is interacting both with its DNA
site and with protein A? If B did dissociate from its DNA site, it would still be held in close approximation to that
site because of its interaction with the bound protein A. Its effective concentration goes up and you should readily
image that it would rebind very quickly to its DNA site. The net effect would be that it's apparent koff
would decrease, which would increase its apparent binding affinity and decrease its apparent Kd (remember that Kd = koff/kon).
Hence prior binding of A would lead to cooperative binding of protein B.
EPIGENTIC CONTROL OF DNA TRANSCRIPTION - METHYLATION OF DNA
The fertilized egg is a totipotent cell. That is, through a series of divisions, its progeny cells
can eventually become any of about 200 histologically different cell types. With subsequent cell division in the developing
embryo, cells find themselves in different topological environments and have different cell-cell contact. Through signal
transduction through the cell membrane, these cells start to become different, or differentiate, into other cells types.
They do so by activating and inhibiting the expression of a different set of genes to form a different set of proteins in
the cells. Most cells become terminally differentiated and eventually (after maybe a hundred cell divisions) lose the
ability to divide and hence begin to die. However, a few types of cells, called stem cells, retain the ability
to differentiate into other cells types in a regenerative process. These cells are pluripotent in that they can
differentiate into other cell types.
How do dividing cells know what types of genes to actively transcribe? How can they have "memory"
of the cells type they were before division? This appears to happen without alteration of the nucleotide sequence of
the DNA in these cells. The main mechanism appears to be a inheritable but modifiable pattern of chemical modifications
to the DNA (not unlike co- or post-translational modification of proteins) involving methylation/demethylation (by a methylase
and demethylase) of cytosine in CpG dinucleotide repeats in the DNA.
Also proteins can bind to DNA and methylated DNA to modify the course of gene expression in daughter cells. Such chemical
modifications to the DNA which modify gene transcription are examples of epigenetic mechanisms controlling gene expression.
You are all familiar with the cloning of animals from the DNA of adult cells (from Jurassic Park to the
actual cloning of the sheep Dolly). In this process, the nucleus from an adult somatic cell (like a check epithelial
cell) is removed and placed in an egg from which the nucleus has been removed (enucleated). The egg now has a full complement
of DNA, just like an adult cell, but it didn't get the full set by normal means - i.e. by receiving half from a sperm to complement
its own normal half. There is another potentially big problem. The DNA in the egg has the methylation pattern
of a terminally differentiated adult cell. It must be reprogrammed by undergoing extensive demethylation and remethylation
to the "correct" epigenetic methylation state if the egg has a chance to form a normal embryo, fetus, and adult. Obviously
this can happen, as evidence by Dolly and success in cloning cows, cats, pigs, and mice. However, it is very difficult
to achieve and probably accounts for the low success rate of cloning.
Methylation patterns can account for gene silencing (in which one gene in a pair of identical chromosomes
is not expressed) and inactivation of one entire X chromosome in a female (who has 2 X chromosomes). In general,
transcription from genes that are methylated is inhibited.
CHROMATIN REMODELING AND GENE EXPRESSION
Control of DNA transcription in eukaryotes was thought to involve the assembly of many proteins at
the promoter into a pre-initiation complex (PIC). Once assembled, RNA polymerase could bind and transcription
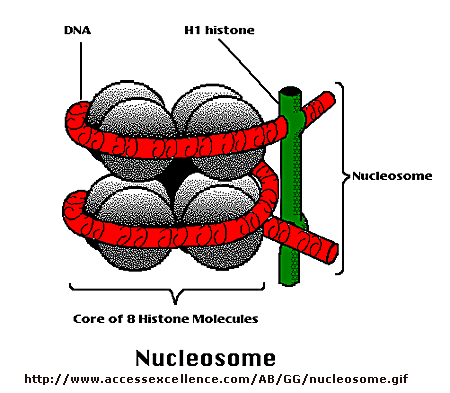
would be initiated. But wait a minutes! Isn't DNA packaged in the nucleus into chromatin in which 147 BP of DNA is wound around a core of 4 pairs of positively charged histone proteins - including H2A,
2B, 3, and 4 - to form a nucleosome, seen under a microscope as beads on a string?
Isn't this chromatin further wound into fibers which result in the classic picture of sister chromatids
ready to separate at cell division. How could the transcription factors and RNA polymerase recognize target sites on
DNA given this degree of "folding" and condensation of the DNA?
Clearly the complex compacted state of DNA and its interaction with the histone proteins must be "remodeled"
to allow interactions of the transcription factors and RNA polymerase (which is about the same size as a nucleosome).
The regulation of this chromatin remodeling clearly affects gene transcription, and is another example of epigenetic changes
that can affects phenotype. The state of chromatin structure is regulated by enzymes that affect histone structure and
function by chemically modifying the histone proteins (through acetylation, methylation, and phosphorylation) .
Likewise, the DNA at the promoter region is changed by enzymes that remodel the DNA through an ATP dependent series of modifications.
For example when histones are modified by histone acetyltransferase (HAT's), other modeling factors (SWI/SNF)
are recruited to the chromatin. Chromatin remodeling would also be affected by that cell cycle stage of the cell.
For example, chromatin condensed in sister chromatids ready for cells division would have different remodeling requirements
for gene transcription than might chromatin in the form of bead on a string. Likewise remodeling efforts would also
be gene-specific.
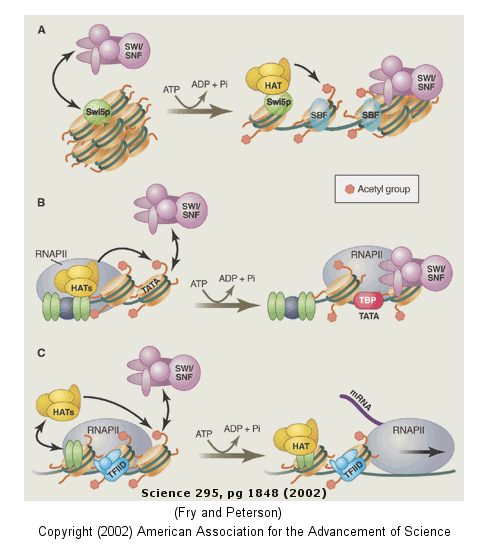
The figure below shows how remodeling is coupled to formation of the pre-initiation complex
for three genes:
-
yeast HO gene: Swi5p activator binding results in the interaction of the SWI/SNF
ATP-dependent remodeling enzyme, which leads to the binding of histone acetyltransferase (HAT). These facilitate
formation of the pre-initiation complex.
-
human interferon-b gene: gene sequences known
as activators, 5' to the promote, bind HATs. When histones are acetylated, SWI/SNF interacts to remodel
the chromatin and facilitate PIC formation.
-
human a-1 antitrypsin gene: the PIC
is preformed and recruits HAT and SWI/SNF, which leads to gene transcription.
Alternations in chromatin remodeling could lead to changes in gene expression, in some cases
causing cancer. SNF5 is a component of the SWI/SNF complex and in its normal form acts to suppress tumors (i.e. its
gene is a tumor suppressor gene). Mutations in SNF5 are associated with rare and aggressive childhood tumors.
Stuart Orkin has developed a technique to alter the gene in some mouse cells to produce an inverted gene which produces no
functional SNF5. Cells with this mutation become tumor cells almost immediately.
Figure: Remodeling of Chromatin and Control of DNA Transcription
DNA winds around the histone core to form the nucleosome. However, histone tails not associated
with DNA binding protrude from the nucleosome, and the function of these tails is just being unraveled. The amino acids
in these tails are clearly sites for posttranslational modifications, including methylation, acetylation, and phosphorylation.
When modified, these tails would provide additional binding sites for protein which could regulate transcription and
chromatin modeling, thus modifying the "genetic code". Understanding the "histone code" and how it affects gene transcription
becomes important. For example, the methylation of Lys 9 on histone 3 leads to binding of heterochromatin-associated
protein, leading to inhibition of gene transcription (an example of epigentic silencing). Acetylation of
the tails generally leads to activation of gene transcription at that site.
Epigenetic changes (through methylation of DNA or acetylation, methylation, and phosphorylation
of histone proteins causing chromatin remodeling may change phenotype (characteristics of the individual) as evidenced by
the fact that identical twins can eventually diverge in ways that effect their propensities to disease. Differences
in diet and lifestyle, which can alter disease propensity, might exert their effects through epigenetic changes in gene expression.
The Human Epigenome Consortium is developing a catalog of methylation pattern differences in the human genome which might be correlated with disease risk.
CONTROL OF GENE EXPRESSION BY RNA MOLECULES
What accounts for the increased complexity of organisms like humans? As was discussed in the DNA
chapter, it is not the number of chromosomes or even the number of possible genes in an organisms. One big difference
between bacterial and human cells, for example, is the percentage of DNA coding for proteins. In bacteria, most of the
DNA codes for proteins, but in human eukaryotic cells, most of the DNA (up to 98%) is "junk" in that it does not code for
proteins. The DNA consists of intervening sequences within DNA coding for a given protein, and sequences between genes.
Up to 98 % of the RNA transcribed in human cells is derived from this "junk" DNA. What function does this RNA serve?
New evidence shows that this RNA binds to other RNA molecules like mRNA (to inhibit its translation), to DNA (to control gene
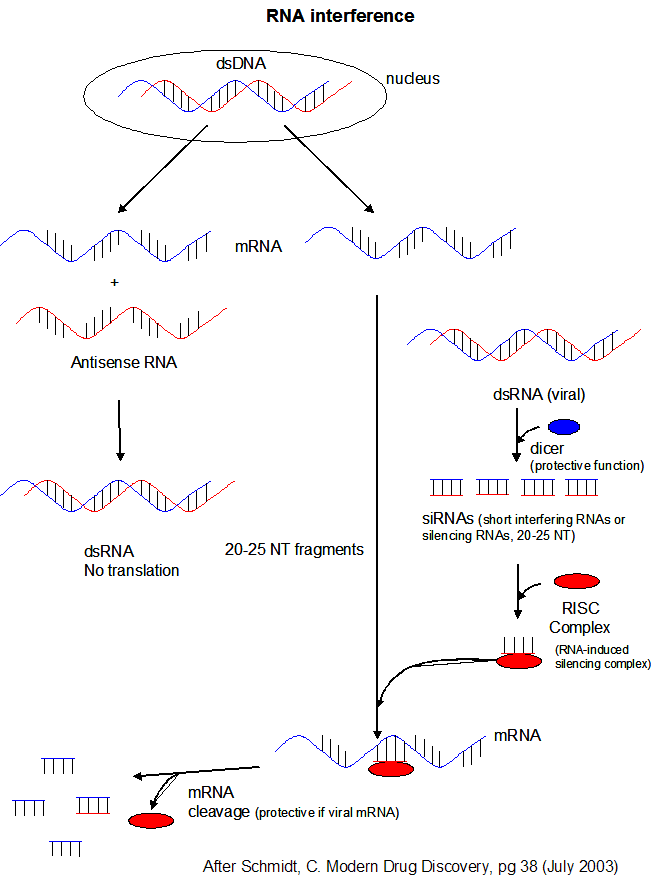
transcription) or to proteins (to alter gene transcription as well). These process are called RNA interference (RNAi)
MicroRNAs (miRNAs) are formed when an enzyme called dicer cleaves a host RNA gene transcript that in animal cells contains a stem-loop hairpin structure. Dicer recognizes the
double-stranded regions of the stem. The cleaved RNA might then bind a host mRNA, inhibiting its translation.
If the complementarity of the mRNA and miRNA is less than ideal, then binding of miRNA to the mRNA may only attenuate the
translation of the mRNA..
Another class of RNA, short interfering
or silencing RNAs (siRNAs) can also
infer with mRNA translation. siRNAs are formed when dicer cleaves viral double-stranded RNA in infected
cells. (Viruses often produced dsRNA during their life cycle). These differ from miRNA in that siRNA are
not derived by transcription from discrete host gene. Dicer works by cleaving the dsRNA to form a small dsRNA
between 20 and 25 nucleotide pairs long called siRNAs. These can then bind to a protein
complex called RISC (RNA-induced silencing complex), which promotes unraveling of the siRNA.
A complex of RISC and one of the short RNA strands from the siRNA then can bind to complementary stretches in mRNA for a specific
gene (viral or host) and inhibit translation of the mRNA into protein. Inhibition occurs when RISC complex cleaves
the RISC-siRNA- mRNA complex. (i.e one of the components in the RISC complex is an RNA nuclease.) This mechanism
might be linked to defense mechanisms of virally-infected cells by inhibition of viral mRNA translation.
siRNAs can also bind to host RNA targets by binding to host mRNA of complementary sequence to form a dsRNA
complex, inhibiting its translation. This technique has recently been used to ascertain the functions of gene products
in the nematode worm, C. elegans. This organism has about 20,000 genes which code for proteins. Kamathk
et. al. have fed these worms E. Coli transformed with plasmid DNA designed to produced dsRNA upon transcription, one
strand of which was complementary to mRNA sequences in the worm. Plasmids containing almost 17,000 different dsRNA encoding
genes were constructed and used to "knock out" gene expression (by forming dsRNA complexes from the mRNA) by RNAi. Phenotypic
changes in the organism were studied. About 1700 of the dsRNA experiments led to observable (phenotypical) changes in
the organism. Genes whose inactivation was lethal (and hence were essential for survival) were generally those that
had counterparts in all other organism, while those associated with nonlethal changes were more likely to be homologous to
higher organisms and more recently evolved. They also selectively looked at which genes influenced lipid metabolism
by incorporating a fluorescent type which bound to lipid deposits in the organism. Around 300 genes were found to influence
fluorescence and hence regulate fat deposition in the organism.
Figure: RNA Interferene: Antisense and Silencing
Recently, a new mechanism in control of gene expression has been offered which involves regulation of
translation of a mRNA. mRNA must have a sequence, the Shine-Delgarno sequence, which allows it to bind to ribosomes.
If a ligand binds to this site, mRNA could not bind to the ribosome and translation would be inhibited. Such is the
case in the mRNA encoding proteins involved in the transport and synthesis of vitamins B1 (thiamine) and B12 (adenosyl cobalamin).
Thiamin and thyamine pyrophosphate were shown to bind to the leader sequence of an E. Coli mRNA involved in thiamine
biosynthesis and inhibit the translation of the mRNA. This allosteric mechanism for inhibition makes physiological sense
since the presence of high levels of cellular B1 would obviate the need for its synthesis or transport.
EUKARYOTIC SPECIES COMPLEXITY AND CONTROL OF GENE TRANSCRIPTON
The increasing complexity of eukaryotic organisms was thought to arise from an increasing number of genes.
This simplistic assumptions has not been validated from the results of sequencing and annotating the genomes of many eukaryotic
organisms. Compare these statistics: the number of putative genes in the simple nematode round worm C. Elegans,
the fruit fly drosophila, and the human are approximately 20,000, 14,000, and about 36,000. There seems to be little
correlation of species complexity with number of genes. Other possible mechanisms for increasing complexity from a given
genome size include producing different proteins from the same genes through differential splicing of RNA transcripts and
rearranging DNA as occurs in immune cells to produce the huge repertoire of possible antibody molecules necessary for recognition
of nonself molecules (such as viruses and bacteria). These mechanisms can not account for the incredible complexity
of the human species. Levine and Tjian have proposed two other mechanisms that could account for increasing complexity.
Complexity would arise from the number of gene expression patterns and involve the involvement of nonprotein-coding regions
of the genome, which in humans accounts for up to 98% of the genome. One mechanism requires the present of greater number
and complexity of DNA regulatory sequences (enhancers, silencers, promoters) in more complex organisms. Since these
sequences are in the DNA (the molecule that is transcribed), they are called cis-regulatory sequences. The second
mechanism involves an increase in the elaboration and complexity of proteins (trans-regulatory elements) that regulate
gene expression in more complex organisms.. These proteins could include transcription factors, proteins interacting
with enhancer sequences, and proteins involved in chromatin remodeling (described above). They estimate that up to a
third of the human genome (1 billion base pairs) might be involved in the regulation of gene transcription. In addition,
5-10% of all proteins expressed from genes appear to regulate gene transcription. There appears to be about 300,
1000, and 3000 transcription factor in yeast, drosophila and C. elegans, and humans, respectively. There is about
one transcription factor for every gene in yeast, but one for every ten in humans.
In simple eukaryotes, cis regulatory elements would include the promoter (TATA box region), and upstream regulatory sequences
(enhancer) and silencers about 100-200 base pairs from the promoter. In more complex eukaryotic species like humans,
the promoter is more complex, containing the TATA box, initiator sequences (INR) and downstream promoter elements (DPE).
Upstream cis regulatory elements (as far as 10 kb from the promoter) include multiple enhancers, silencers, and insulators.
Most promoters have TATA boxes, where TATA Binding Protein (TBP) binds. Upstreams elements in turn regulate the binding
of TBP.
Comparative Genomics - Gene Expression Differences Between Humans and Chimpanzees
Our closest biological relative is the chimpanzee, who branched off from a common ancestor of both of
us about six million years ago. Our DNA sequence appears to be 98.6 % identical (not just homologous). If we are
so close in our genetic blue print, how can we be so different. They are many possible conjectures that may soon be
answered when the chimp genome is sequenced and compared to that of the human. Our genes are presumably very similar.
People suspect that there are two major kinds of differences that make our species different:
- our genes are very similar but are transcribed differently in the two species. Recent evidence
show that the types of RNA transcribed by human and chimp livers are very similar, but many more genes are transcribed in
human brains compared to chimp brains.
- humans may have lost genes (or their function) that are required for chimp survival in the jungle.
The observations that chimps are resistant to many of the disease pathogens that affect humans (immunodeficiency viruses
like HIV, influenza A virus, hepatitis B/C, malarial parasite) could be explained by the loss of "protective" genes
in humans. In addition, cardiovascular disease and certain types of cancer are rarer in chimps. Humans have apparently
"lost" genes involved in body hair, strength, and early maturation, traits that would adapt the chimp to life in the jungle.
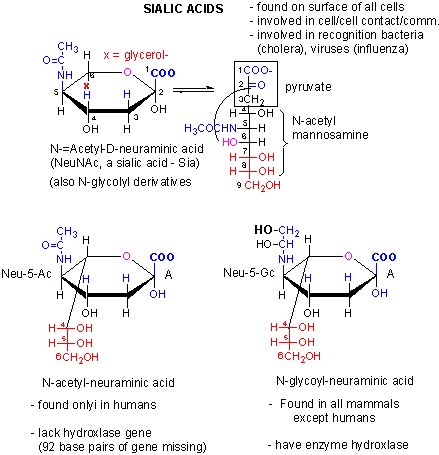
We previously discussed an example of a loss of gene function in humans. We have lost a hydroxylase gene involved in formation of certain types of sialic acids, specifically N-glycolylneuraminic acid, found
on cell surface glycogroteins of mammals other than humans. Chimps have a lectin receptor for this sialic acid. Recent
work has shown that human lack a critical Arg in our version of the lectin that would recognize N-glycolylneuraminic
acid, making it unable to bind this ligand. Hence both pairs of genes involved in these type of interactions
(cell:cell) are missing. Since sialic acid molecules are often involved in pathogen:host binding, these difference in
humans compared to chimps might account for the difference in disease susceptibility as mentioned above.
With respect to gene transcription in the brain, Lai et al. have found a mutation in the human gene FOXP2,
a transcription factor, in a family that has significant difficulty in controlling muscles required for articulation of words.
This mutation also causes problems in language processing and grammar construction. Comparsion of the normal human gene
with other primate genes shows distinct differences in the human gene which may have conferred on human the ability to use
speech.
{kind=link}
{kind=link}
{kind=link}