|
INTRODUCTION
Given the number of possibly nonnative states, it is amazing that proteins fold to the native state at
all, let alone in a reasonable time frame. Consider this greatly simplified view of protein folding for a protein containing
100 amino acids. If each amino acid can adopt only 3 possible conformations, the total number of conformations could
be 3100 = 5 x 1047. Assuming that it would take 10-13s to change each conformation,
the time required to "test" all conformations would be 5 x 1034s or 1027 years, longer than the age
of the universe (14 x 109 yr). Yet the protein can fold within seconds. This paradox is called the Levinthal
paradox, after Cyrus Levinthal.
Lubert Stryer (in his classic Biochemistry text), shows a way out of this dilemma by using an analogy
of a monkey sitting at a typewriter, and typing this line out of Hamlet: "Me thinks it is like a weasel." Random
typing would produce that line after 1040 keystrokes on average, but if the correct letters were maintained,
the number of keystrokes would be in the realm of a few thousand. Proteins could fold more quickly if they retain native-like
intermediates along the way. Also remember that much of conformation space is already restricted by allowed phi/psi
angles. (Remember all white area in the Ramachandran plot?)
Before we study the classic experiment of protein folding conducted by Anfinsen, study the simpler analogy
below:
Figure: Socks and protein folding
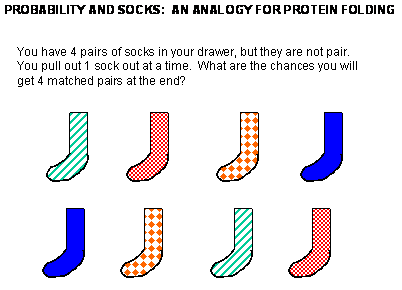
The classic experiment of Anfinsen has shown that, at least for some proteins, all the necessary
and sufficient information required to direct the folding of a protein into the native state is present in the primary sequence
of a protein. Anfinsen studied the in vitro (outside the cell, as opposed to in vivo, which is inside the
cell, tissue, organ) folding of a single chain protein, RNase, which has 4 intrachain disulfide bonds. He unfolded,
or denatured, the protein to the nonnative or denatured state with 8M urea, in the presence of the reducing agent,
b-mercaptoethanol, which reduces the disulfide
bonds to free Cys residues. He then removed the bME
using dialysis, allowing the disulfides to reform. Next he removed the denaturing reagent, urea. To monitor if the protein
was correctly refolded or renatured, he tested the activity of the protein compared to native protein. He found that the "refolded"
protein retained only 1% of its initial activity. If, however, he added a catalytic amounts of bME, the protein soon retained 100% of its initial activity. For his work, he was awarded the
Nobel Prize in Chemistry in 1972.
Figure: Anfinsen Experiment: Folding of RNase
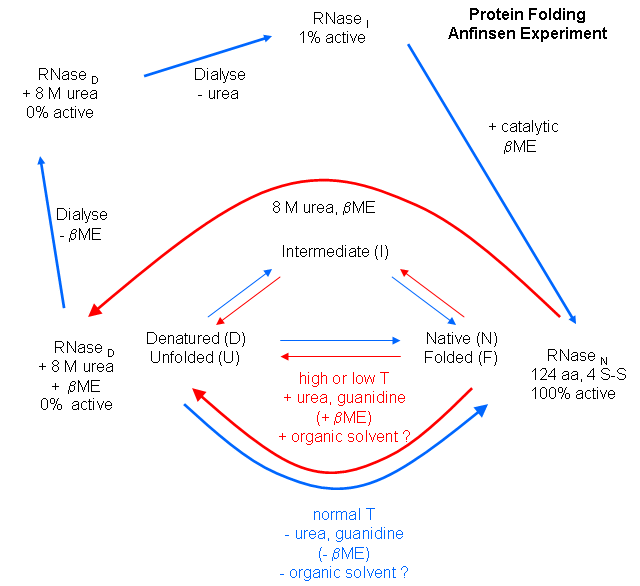
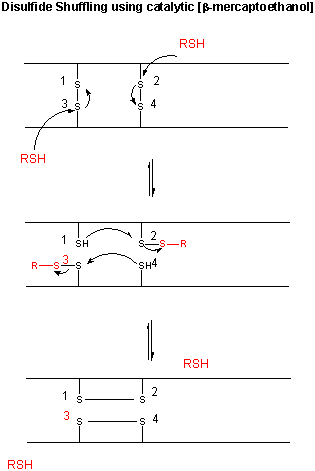
Figure: CATALYTIC SHUFFLING OF DISULFIDES WITH BETA-MERCAPTOETHANOL
Scientists have investigated the folding of proteins both in
vitro and in vivo. In vitro experiments involve denaturing the protein with urea, guanidine hydrochloride,
or heat, then refolding the protein by removing the perturbant (denaturing agent), using spectral techniques to follow the
process. In vivo experiments involve the study of intracellular proteins that assist folding. The in vitro experiments
involve unfolding the native state and then refolding it, while the in vivo ones involve folding of the newly synthesize
protein. An understanding of protein folding can not be separated from an understanding of protein stability, and an
understanding of the nature of the native and denatured state.
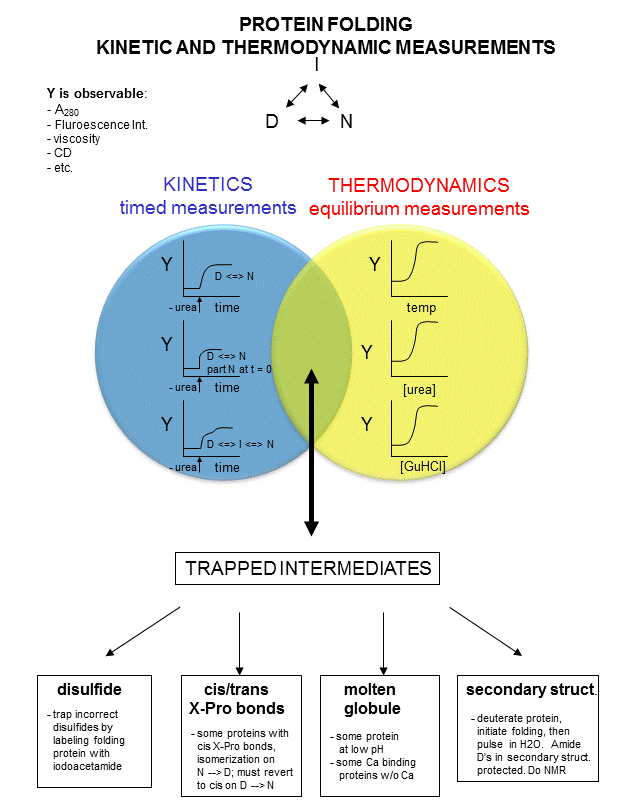
In studying protein folding and stability/structure of the native and denatured states,
both equilibrium (thermodynamic) and timed (kinetic) measurements are made. Folding occurs in the ms to second range, which
limits the ability to study the presence of intermediates in the process. Some clever methods have been developed to study
intermediates in protein folding by trapping specific intermediate structures, and investigating their structure and stability
in a "leisurely" fashion. Alternatively, intermediates can be studied as they occur using stop flow kinetic measures. In this technique, a protein under denaturing conditions is rapidly mixed with a solution containing no denaturant
or protein by injecting both solutions into a mixer/cuvette using syringes. The denaturant in the protein solution is
now diluted such that renaturation can occur. Spectral measurements can begin at once. A diagram summarizing these
methods is shown below. Study it in conjunction with the text which follows.
Figure: Kinetic and thermodynamic measurements of proteins stability and folding
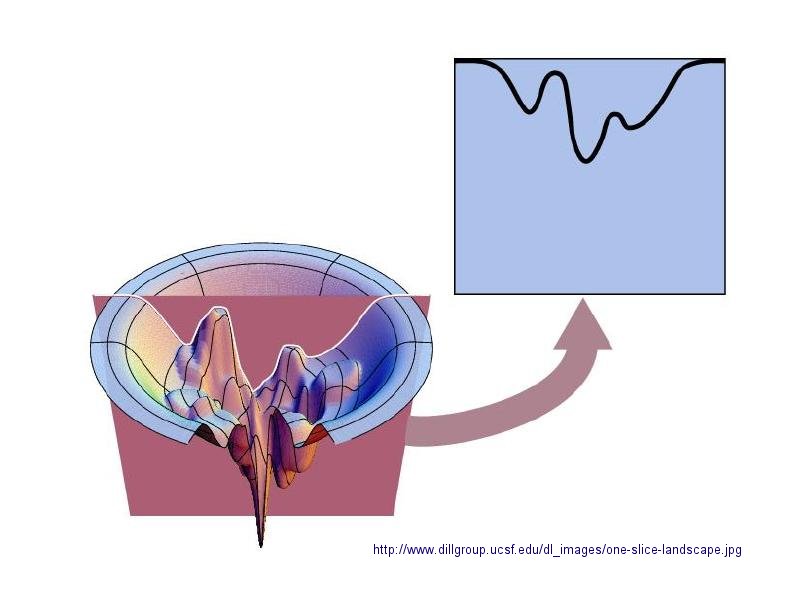
In considering the folding pathway, we will consider that the native protein represents the global energy minimum.
All other states represent variations of the denatured state. Some, closer in energy to the native state, could be considered
intermediates in the folding process. Instead of considering a folding "trajectory", consider protein folding occurring
within a large folding landscape of free energy. Folding appears to proceed not by an obligatory pathway but a probablistic
or stochastic search of possible conformation. The free energy landscape must be shaped somewhat like a funnel such that a
proteins could adopt a "reasonable" number of conformations which lead to the native state. Evolution has surely selected
for sequences that can make it to that state. Localized secondary structure motifs (like a short alpha helix and beta
turns) can form quickly (about 1 ms). Small proteins folding occurs, depending on the structure,
over a wide time frame (ms to minutes). Mostly likely, a small number of amino acids coalesce into a core which nucleates
folding into structures that are similar to the native state. Finally packing interactions collapse the structure into
the native state.
In general, the more complex the fold of the backbone, the longer it takes the protein to fold. If complexity
requires more interactions among distal regions of the polypeptide change, then the more complex the fold, the less probable
that random interactions would lead to quick protein folding. The mechanisms of folding for larger proteins (greater
than 100 amino acids) appear to proceed through intermediates, suggesting that different domains of the protein can fold independently.
Figure: Protein Folding Landscape: One View from Ken Dill
In Vitro Protein Folding
Early studies of protein folding involved small proteins which could be denatured and refolded
in a reversible fashion. A two state model, D <===> N, was assumed. The denaturants were heat, urea, or guanidine HCl.
Since the denatured states are less compact than the native state, the viscosity of the solution can be used as a measure
of denaturation/renaturation. Likewise, the amino acid side chains in the differing states would be in different environments.
The aromatic amino acid Trp, Phe, and Tyr absorb UV light. After excitation, the electrons decay to the ground state through
several processes. Some vibrational relaxation occurs, bringing the electrons to lower vibrational energy levels. Some of
the electrons can then fall to various vibrational levels at lower principle energy states through a radiative process. The
photons emitted are lower in energy and hence longer in wavelength. The emitted light is termed fluorescence. The wavelength
of maximum fluorescent intensity and the lifetime of the fluorescence decay is very sensitive to the environment of the amino
acids. Hence fluorescence can also be used to measure changes in protein conformation. Other spectral techniques like CD spectroscopy
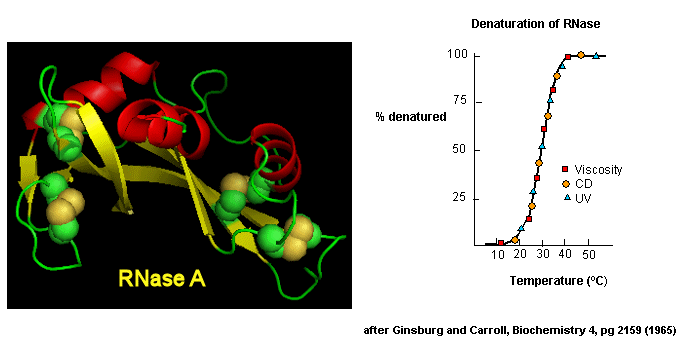
as well as simple absorbance measurements, are used. For small, single domain proteins (such as RNase) undergoing reversible denatuation, graphs showing the extent of denaturation using
each technique above, are superimposable, giving strong validity to the two state model.
Figure: Reversible denaturation
This simple model needed to be expanded as more proteins were studied. Some intermediates
in the process were detected.
- Some proteins show two steps, one slow, one quick, in refolding studies, suggesting an
intermediate. The longer a protein is kept in the denatured state, the more likely it is to display an intermediate. One accepted
explanation for this phenomena is that during an extended time in the D state, some X-Pro bonds might isomerize from trans
to the cis state, to form an intermediate. Alternatively, as in the case of RNase, which has a cis X-Pro bond in the native
state, denaturation causes an isomerization to the trans state. In the case of RNase, to refold, the accumulating intermediate
I must reisomerize in a slow step to the cis state, followed by a quick return to the N state.
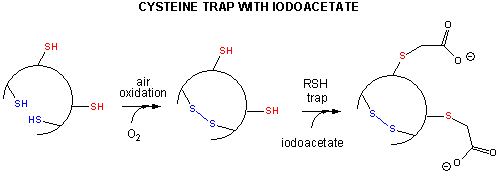
- Some proteins which contain multiple disulfide bonds that must reform correctly after reductive
denaturation can refold into intermediates with the wrong S-S partner. Such intermediates can be trapped by stopping further
S-S formation during refolding with the addition of iodoacetamide.
Figure: addition of iodoacetamide
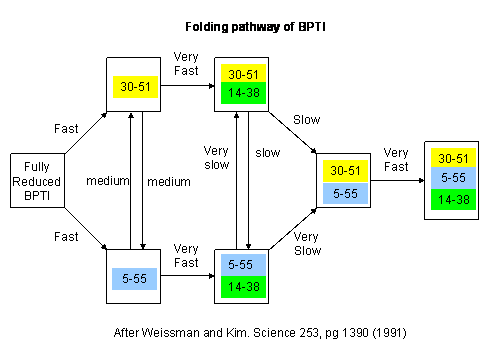
As an example consider the following data on bovine pancreatic trypsin inhibitor.
Figure: Bovine pancreatic trypsin inhbitor (BPTI): Folding Kinetics
- only native disulfide structures seem to form.
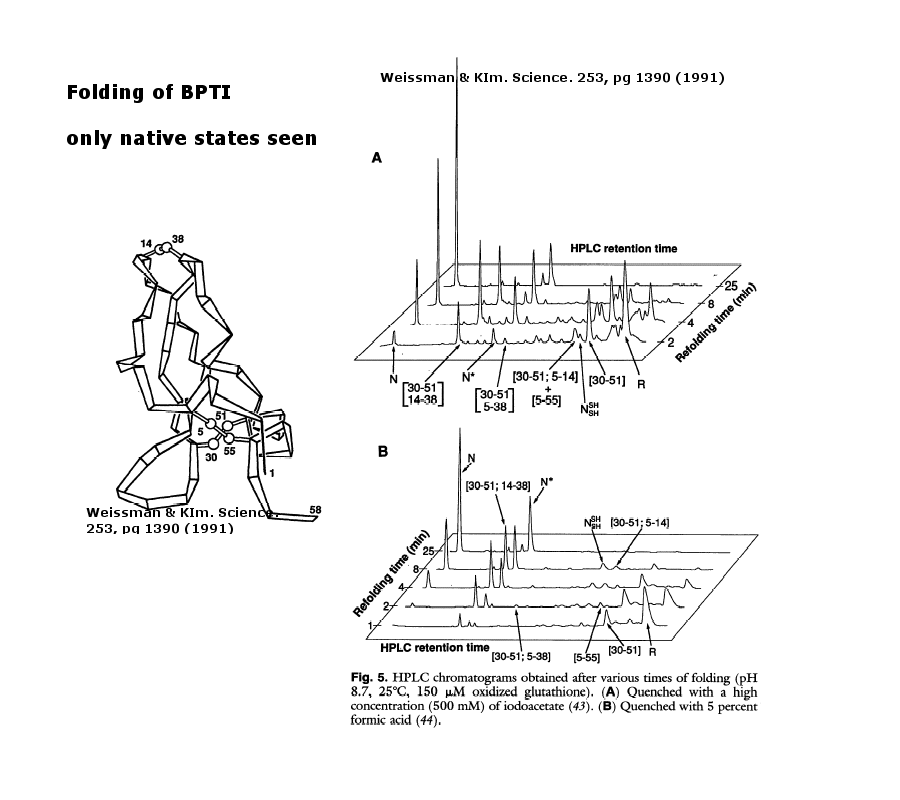
Figure: BPTI Folding Pathway In Vitro - gives possible scheme
of folding intermediates
- Some proteins form partially folded but stable intermediates when folded under partially
denaturing conditions. A good example is lactalbumin, which under mildly acidic conditions (pH 4), low levels of guanidine HCl, or neutral
pH and low ionic strength in the absence of calcium (which normally binds to the protein), forms a stable, isolatable
intermediate (I) called the molten globule (MG).
Figure: lactalbumin
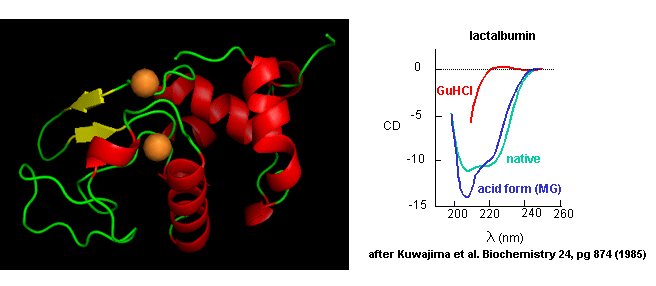
Data show that the MG is about 50% larger in volume than the N state. This compares to the denatured
state, which can be 300% larger than the native state. Hence, it is more like the native state as studied by hydrodynamic
techniques, but with more solvent accessibility of hydrophobic side chains. The MG has a similar CD spectra as the native
state, but the aromatic side chains display the same UV absorption and fluorescent characteristics as the protein in 6 M guanidine
HCl, suggesting that the final tertiary state has not yet completely formed. The secondary structure in the MG may not
be the same as in the native state.
NMR techiques can also be used to detect folding intermediates. Using this technique,
proteins are unfolded in D2O, which will cause the exchange of all C's with ionizable protons,
including, the amide H's .
Figure: Exchange of all Cs with ionizable protons, including the amide Hs
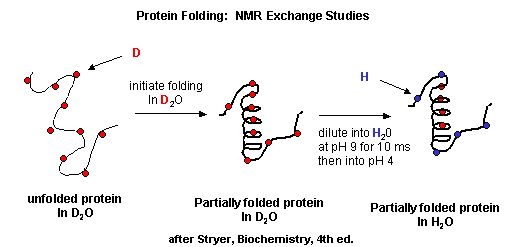
Refolding is initiated by diluting the protein into a solution without the denaturatant,
but still in D2O. As the protein folds and becomes more compact, the buried atoms are now sequestered from the
solvent, and no longer readily exchange Ds. Then the protein is placed in H2O at pH 9.0 for 10 ms, after which
the pH is changed to pH 4.0. D --> H exchange is promoted at high pH, and quenched for the amide Ds and Hs at low pH.
Amide H's that continue to exchange must be accessible to water. Those that aren't are usually buried in secondary structure.
Figure: Experimental data on
model proteins. How would you interpret these
graphs.
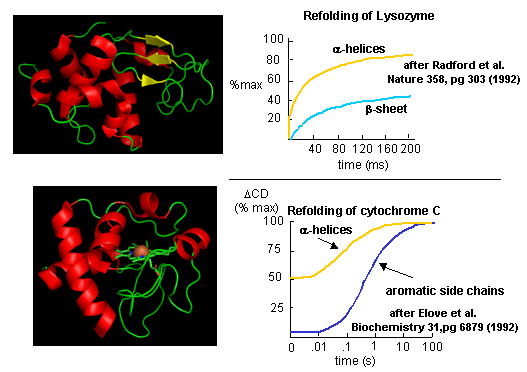
When the same techniques are applied to large, multidomain or oligomeric proteins, only
a few percenage refold in vitro. Incorrect intermolecular interactions and heterogeneous aggregation seems to be the main
problems which prevent correct protein folding in vitro.
In Vivo Protein Folding
There are many differences between how a protein might fold or unfold in a cell compared
to a test tube.
- The total concentration of all the proteins and nucleic acids in cells are estimated to
be about 350 g/L, or 350 mg/ml. Most measurement in the lab are conducted in the range of 0.1 to 10 mg/ml
- Proteins are synthesized in cells from an N to C terminal direction. Hence the nascent
protein, as it emerges from its site of synthesis (the ribosome), might fold into intermediate structures since not all of
the protein sequence is yet available to direct folding.
- Proteins are synthesized in the cytoplasm, but they have to find their final place in the
cell. Some end up in membranes, some must translocate across one or even two different membranes to end up in specific organelles
like the Golgi, mitochondria, chloroplasts (in plant cells), nuclei, lysosomes, peroxisomes, etc. Do they translocate in their
native state?
Additional evidence suggests that protein folding/translocation requires assistance (i.e.
catalysis) in the cell.
- Mutant cells defective in certain proteins can lead to the accumulation in the cells of
misfolded and aggregated proteins.
- eukaryotic genes (taken from higher cells which contain nuclei and internal organelles),
when transferred into prokaryotes (bacteria, like E. Coli), can be expressed to form protein, but they often misfold
and aggregate in the bacterial cells and form structures called inclusion bodies.
Hence recombinant proteins expressed in vivo have the same problems in folding as
larger proteins in vitro. In both cases, conditions favor accumulation of nonnative proteins with exposed hydrophobic
groups leading to aggregation. Aggregation also occurs in vivo when a protein is over-expressed or expressed
at a higher temperature than normal. Why? Mutant cells have been selected that actually suppress inclusion bodies
in vivo. This effect was mediated by a class of proteins which are expressed by the bacteria and other cells when their
temperature is raised. The function of these proteins, called heat shock proteins (Hsp), was unknown until it was realized
that they facilitate correct protein folding, in part, by binding to denatured proteins in the cells before they aggregate
into inclusion bodies. Further studies discovered a large number of proteins that seem to facilitate protein folding and prevent
aggregation in vivo. These proteins are now called molecular chaperones. They are classified
on the basis of their molecular weight) and can be divided into at least two families.
Hsp-70 Family (including DnaK/DnaJ and GrpE proteins)
. Examples include the immunolglobulin heavy chain
binding protein (BiP) and alpha crystallin, which comprises 30% of the lens proteins in the eye, where it functions, in part,
to prevent nonspecific, irreversible aggregates. These proteins (70K MW):
- bind to growing polypeptide chains as they are synthesized on ribosomes.
- express activity as monomers.
- have ATPase activity - i.e. they cleave the phosphoanhydride ATP (which can drive reactions).
- bind short, extended peptides, which stimulates the ATPase activity
- release bound peptides after ATP cleavage
Chaperonins - including chaperonin 60 (or GroEL in
E. Coli) and chaperonin 10 (or GroES in E. Coli) in chloroplasts, mitochondria and bacteria, and TCP-1 in eukaryotic cytoplasm.
These proteins:
- bind to proteins after they have left the ribosome or have been transported into organelles
like mitochondria.
- express activity as multimers. GroEL consist of two stacks of rings of monomers, with 7
monomers in each ring (each monomer around 60K MW), forming a hollow cylinder. GroES consist of one single ring of 7
monomers (each 10K MW). The GroES complex forms a lid over one open end of the GroEL cylinder. Proteins can fold
within the cavity in GroEL (lined with hydrophobic patches) without "fear" of aggregation. GroEL also binds
and cleaves ATP, leading to conformational changes inside the barrel and hiding of the hydrophobic patches in Gro EL, which
leads to the releases of the unfolded peptide. The process proceeds until the folding protein passes through the barrel
and is released in its correct folded state.
- bind nonnative proteins at the GroEL opening of a complex of GroEL and GroES, which has
a large hydrophobic cavity.
- A Review of Molecular Chaperonins in Disease
- Chaperonin Home Page
GroEL has also been shown to bind in its hydrophobic cavity a fluorescent CdS semiconductor nanoparticle
which can be released on addition and cleavage of ATP. There are two classes of chaperonins:
- Class I: Those found in bacteria, chloroplasts and mitochondria. The have structures analogous
to GroEL (two rings of 7 identical monomers) and Gro ES.
- Class II: Those found in archebacteria and in the cytoplasm of eukaryotic cells. These contain
two rings of 8-9 subunits which may not be identical.
Other chaperons have proven to be of clinical significance. Hsp 90 is a chaperone that is expressed
both in normal and tumor cells. It appears to have special importance in tumor cells in helping key proteins involved
in malignancy (signal transduction proteins such as HER-2/ErbB2, Akt, Raf-1, Bcr-Abl, and p53) to maintain their shapes under
conditions of drug exposure and the inherent genetic instability present in the cells. Drugs that bind to and inhibit
Hsp90 appear to have much greater effect on tumor cells, making this protein a new chemotherapeutic target to treat cancer.
Recent studies by Kamal et al. have shown the drug 17-AAG binds Hsp90 about 100 times as strongly in tumor cells than in normal
cells. Hsp 90 appears to be complexed to other "co-chaperones" in the tumor cells which lead to higher drug binding
affinity. The chaperone complex may actually induce the drug to adopt a different conformation which binds with higher
affinity. A new biotech firm, Conforma Therapeutics, has centered its efforts on designing novel anti-tumor drugs based on chaperone proteins.
 Online Literature: Ranford, J. et al. Chaperonins are cell-signalling proteins: the unfolding biology of molecular chaperones. Expert Reviews in Molecular Medicine ©
Cambridge University Press ISSN 1462-3994 Online Literature: Ranford, J. et al. Chaperonins are cell-signalling proteins: the unfolding biology of molecular chaperones. Expert Reviews in Molecular Medicine ©
Cambridge University Press ISSN 1462-3994
Additional Proteins Which Catalyze Protein Folding: Chaperons
function to minimize protein aggregation, which increases the efficiency of the entire process. Other proteins in the
cell actually catalyze specific steps. Here are two examples:
- Protein Disulfide Isomerase (PDI) - catalyzes the conversion of incorrect to correct disulfides.
The active site consists of 2 sets of the the following sequence - Cys-Gly-His-Cys, in which the pKa of the Cys are much lower
(7.3) than normal (8.5). How would this facilitate disulfide isomerization?
- Peptidyl Prolyl-Isomerase (PPI) - catalyses X-Pro isomerization, by a mechanism which probably involves bending the X-Pro peptide bond. How would this facilitate
the process?
Many proteins have been found to possess PPI activity. One class is the immunophilins. These
are small proteins found in the cytoplasm that bind anti-rejections drugs used to prevent tissue rejection after transplanation.
The immunophilin FK506 binding protein (FKBP) binds FK506 while the protein cyclophilin binds that anti-rejection drug cyclosporin.
The complex of cyclophilin:cyclosporin or FKBP:FK506 binds to an inhibits calcineurin, an important protein (with phosphatase activity) in immune cells (T cells) required for T cell function. In this
case, immunophilin:drug binding to calcineurin inhibits the activity of the T cell, preventing immune attack on the transplanted
tissue, preventing rejection. The immunosuppressant drugs (FK506 and cyclosporin) inhibit the PPI activity of their
respective immunnophilin. The extent to which the PPI activity of cyclophiin is required for its activity is unclear,
but it seems to be important for some of its biological effects.
|