As described in the Introduction to Proteins, we can understand proteins structure at varying level of
complexity.
Figure: Protein Analysis from low to high resolution.
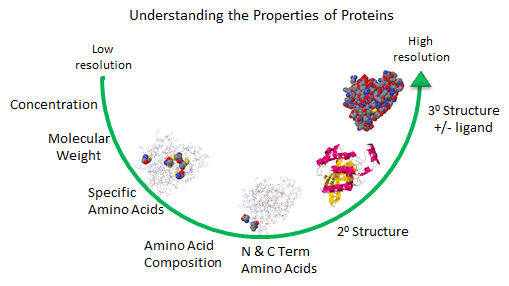
Amino Acid Analysis (Composition)
At a low level of resolution, we can determine the amino acid composition of the protein by hydrolyzing
the protein in 6 N HCl, 100oC, under vacuum for various time intervals. After removing the HCl, the hydrolyzate
is applied to an ion-exchange or hydrophobic interaction column, and the amino acids eluted and quantitated with respect to
known standards. A non naturally- occurring amino acid like norleucine is added in known amounts as an internal standard to
monitor quantitative recovery during the reactions. The separated amino acids are often derivitized with ninhydrin or
phenylisothiocyantate to facilitate their detection. The reaction is usually allowed to procedure for 24, 36, and 48
hours, since amino acids with OH (like ser) are destroyed. A time course allows the concentration of Ser at time t=0
to be extrapolated. Trp is also destroyed during the process. In addition, the amide links in the side chains of Gln
and Asn are hydrolyzed to form Glu and Asp, respectively.
N- and C-Terminal Amino Acid Analysis
The amino acid composition does not give the sequence of the protein. The N-terminus of the protein can
be determined by reacting the protein with fluorodinitrobenzene (FDNB) or dansyl chloride, which reacts with any free amine
in the protein, including the epsilon amino group of lysine. The amino group of the protein is linked to the aromatic ring
of the DNB through an amine and to the dansyl group by a sulfonamide, and are hence stable to hydrolysis. The protein is hydrolyzed
in 6 N HCl, and the amino acids separated by TLC or HPLC. Two spots should result if the protein was a single chain, with
some Lys residues. The labeled amino acid other than Lys is the N-terminal amino acid. The C-terminal amino acid can be determined
by addition of carboxypeptidases, enzymes which cleave amino acids from the C-terminal. A time course must be done to see
which amino acid is released first. N-terminal analysis can also be done as part of sequencing the entire protein as
discussed below (Edman degradation reaction).
Analysis for Specific Amino Acids
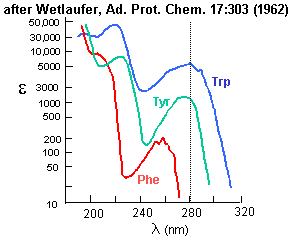
Aromatic amino acids can be detected by their characteristic absorbance profiles. Amino acids with specific
functional groups can be determined by chemical reactions with specific modifying groups, as shown in section 2A.
Figure: amino acid absorbance profiles
Amino Acid Sequence
Two methods exist to determine the entire sequence of a protein. In one, the protein is sequenced; in
the other, the DNA encoding the protein is sequenced, from which the amino acid sequence can be derived. The actually protein
can be sequenced by automated, sequential Edman Degradation.
Figure: Edman Degradation
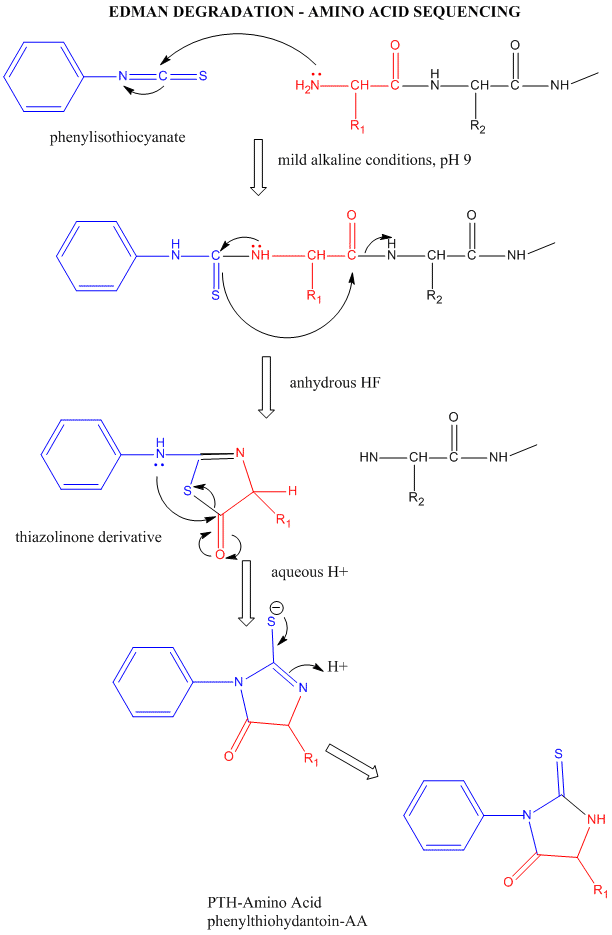
In this technique, a protein adsorbed to a solid phase reacts with phenylisothiocyanate. An intramolecular
cyclization and cleavage of the N-terminal amino acid results, which can be washed from the adsorbed protein and detected
by HPLC analysis. The yields in this technique are close to 100%. However, with time, more chains accumulate in which an N-terminal
amino acid has not been removed. If it is removed on the next step, two amino acids will elute, creating increasing "noise"
in the elution step - i.e. more than 1 amino acid derivative will be detected. Hence the maximal length of the peptide which
can be sequenced is about 50 amino acids. Most proteins are larger than that. Hence, before the protein can be sequenced,
it must be cleaved with specific enzymes called endoproteases which cleave proteins after specific side chains. For example,
trypsin cleaves proteins within a chain after Lys and Arg, while chymotrypsin cleaves after
aromatic amino acids, like Trp, Tyr, and Phe. Chemical cleavage by small molecules can be used as well. Cyanogen
bromide, CNBr, cleaves proteins after methionine side chains. The individual proteins must be cleaved using
two different methods, and each peptide fragment isolated and sequenced. Then the order of the cleaved peptides with known
sequence can be pieced together by comparing the peptide sequences obtained using different cleavage methods.
Many proteins also have disulfide bonds connecting Cys side chains distial to each other in the polypeptide chain. Proteolytic
or chemical cleavage of the protein would lead to the formation of a fragment containing two peptides linked by disulfides.
Edman degration would release two amino acids from such fragments. To avoid this problem, the protein is oxidized with
performic acid, which irreversibly oxidizes free Cys, or Cys-Cys disulfides to cysteic acid residues. A summary of the steps
involved in protein sequencing are shown below:
PROTEIN SEQUENCING STRATEGY - 8 STEPS
- If the protein contains more than one polypeptide chain, the chains are separated and purified.
If disulfide bonds connect two different chains, the S-S bond must be cleaved (as described in step 2) and each peptide independently
purified.
- Intrachain S-S bonds between Cys side chains are cleaved with performic acid. (See above for interchain
S-S bonds).
- The amino acid composition of each chain is determined
- The N-terminal and C-terminal residues are identified.
- Each polypeptide chain is cleaved into smaller fragments, and the amino acid composition and sequence
of each fragment is determined.
- Step 5 is repeated, using a different cleavage procedure to generate a different and overlapping set
of peptide fragments.
- The overall amino acid sequence of the protein is reconstructed from the sequences in overlapping fragments.
- The position of the S-S is located. (See online problem set - Proteins)
A newer method of determining the sequence of a protein uses mass spectrometry. The following description
is derived from: Corthals G.L., Gygi, S.P., Aebersold R. and Patterson, S.D., in Proteome research: 2D gel electrophoresis
and detection methods, Ed. Rabilloud, T., Springer, New York, 1999, pp. 197-231.
"Biological mass spectrometry (MS) is now an indispensable tool for rapid protein
and peptide structural analysis, and the widespread use of MS is a reflection of its ability to solve structural problems
not readily or conclusively determined with conventional techniques. All mass spectrometers (MS) have three essential components
that are required for measuring the mass of individual molecules that have been converted to gas-phase ions prior to detection.
The components are an ion source, a mass analyzer and a detector.
Ions produced in the ion source are separated in the mass analyzer by their
m/z ratio, and (usually) detected by a photomultiplier. MS data is recorded as "spectra" which displays ion intensity versus
the m/z value. The two techniques that have become preferred methods for ionization of peptides and proteins are electrospray
ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI), due to their effective application to a wide range
of proteins and peptides (Fenn et al., 1989; Karas & Hillenkamp, 1988).
Although different combinations of ionization techniques and mass analyzers
exist, MALDI usually uses a time-of-light (TOF) tube as a mass analyzer while ESI is traditionally combined with quadrupole
mass analyzers capable of tandem mass spectrometry (MS/MS). Instruments capable of MS/MS have the ability to select ions of
a particular m/z ratio from a mixture of ions, to fragment selected ions by a process called collision induced dissociation
(CID) and to record the precise masses of the resulting fragment ions. If this process is applied to the analysis a peptide
ions, in principle the amino acid sequence of the peptide can be deduced. "
Recently, MALDI mass spectroscopy has been used to sequence proteins obtained
from fossils. Although most work on sequencing of fossil remains has centered on DNA analyses, some proteins, such as
ostocalcin found in bone, is more stable than DNA, which is susceptible to degradation by ubiquitous enzymes. Osteocalcin
from a 55,000 year old bison bone was identical to that from the modern bison., and was one amino acid different from the
modern cow.
Conformational Analyses of Proteins.
A protein can be considered to have primary, secondary, tertiary, and quaternary structures.
- primary structure: the linear amino acid sequence of a protein
- secondary structure: regular repeating structures arising when hydrogen bonds between the peptide backbone amide
hydrogens and carbonyl oxygens occur at regular intervals within a given linear sequence (strand) of a protein (as in the
alpha helix) or between two adjacent strands (as in beta sheets and reverse turns)
Figure: Secondary Structure
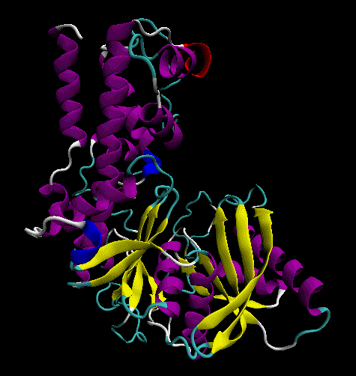
- tertiary structure: the overall three dimensional shape of a protein, often represented by a backbone trace
Figure: tertiary structure
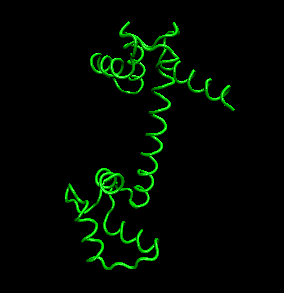
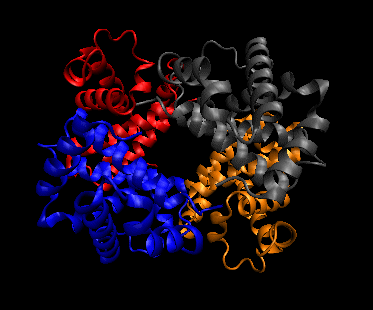
- quaternary structure: oligomeric structure of a multisubunit protein in which separate proteins chains associate
to form dimers, trimers, tetramers, and other oligomers. The different chains in the oligomers may be the same protein
(homooligomers) or a combination of different protein chains (heteroliogomers). The different chains within the oligomer
may be held together by noncovalent intermolecular forces or may also contain covalent interchain disulfides.
Figure: Quaternary structure
2o Structure: The percent and type of secondary structure can
be determined using circular dichroism spectroscopy. In this method, circular (not plane polarized) light illuminates a protein,
which, since it is made of all L-amino acids, is chiral. (The mirror image would be a protein of the same sequence made
of D-amino acids.) Optical activity is observed only when the environment in which a transition occurs is asymmetric. The
peptide bond is asymmetric, and it is this chromophore which undergoes the transition. The electrons are promoted to an excited
state on absorbing light. If the environment of the electrons is dissymetric, as it is in right-handed alpha helices, the
electrons will absorb right and left circularly polarized light differently (for example, they have different molar absorptivities).
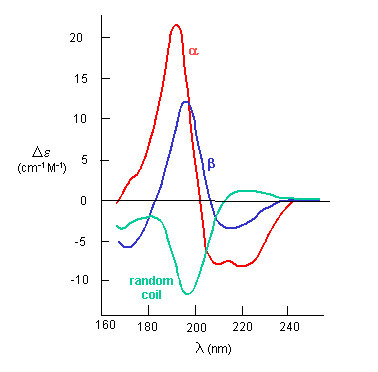
The far-UV CD spectrum of the protein is sensitive to the main chain conformation. The CD spectra of alpha and beta secondary
structure are shown in the figure below.
Figure: The CD Spectra of Alpha-Helix, Beta-Sheet, and Random Coils
3o Structure: Clearly, the highest resolution understanding of protein
structure requires a solution to the 3D structure of the protein. Once that is determined, it is easy to devise computer programs
which will determine what part of the structure is in secondary structure. Three methods are presently useful to determine
the 3D structures of proteins.
A. X-Ray crystallography: If crystals of the protein can be made, traditional x-ray crystallographic
techniques can be used to solve the structure. X-rays irradiate a crystal, which scatters the x-rays, leading to constructive/destructive
interference patterns. Using appropriate math, the interference pattern can be reconverted into the actual structure of the
protein. Check out a fun example showing such a reconstruction of the Parthenon from its diffraction pattern!

These structures are not solution structures, but overwhelming evidence suggest that they do represent
the solution structure. For instance, x-ray structure contain many water molecules which interact with each other and
the protein, a finding expected for a structure that represents the solution structure of the protein. In addition,
substrates and inhibitors can be infused into the crystal and bind with the protein, suggesting again a native-like structure
for the protein in the crystal.
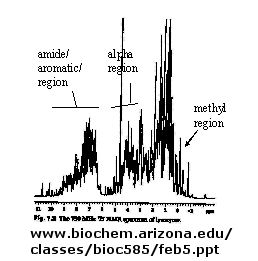
B. NMR: There are many protons in proteins which give a proton NMR spectra. The problem is one
of assignment, since there are so many. Nuclei in different environments absorb energy at different resonant frequencies.
When a proton spin flips, it goes to a higher energy state. It will return to the equilibrium state with some time delay.
Figure: 1D NMR spectra of a protein
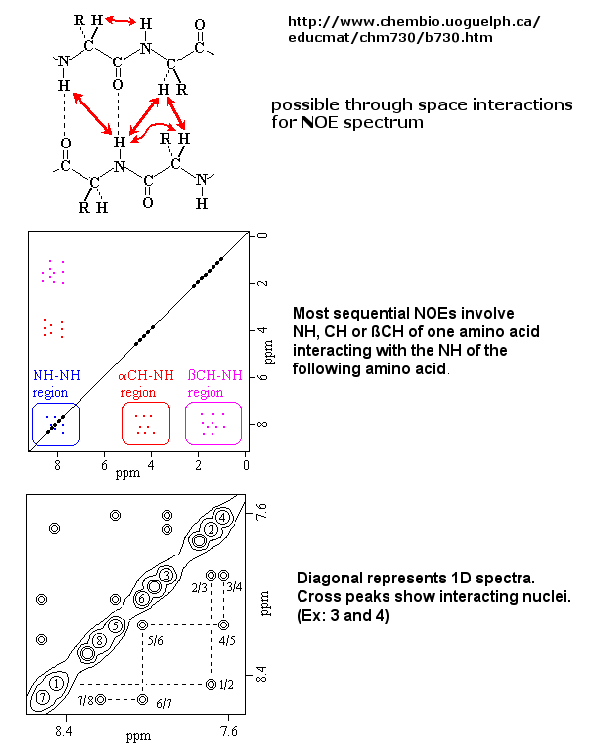
If an unexcited proton is proximal in space, the magnetization can be transferred to the unexcited proton.
This interaction is inversely proportional to the 6th power of the distance between them and is the basis of the Nuclear Overhauser
Effect (NOE). A 2D NOE spectra shows peaks off the diagonal that are correlated, indicated that they are close in 3D space.
Figure: 2D NOSEY spectra of a protein
Multi-dimensional techniques, obtained when isotopes of N(15) and C(13) are present in the protein, can
be used to actually obtain a 3D solution of a small protein. NMR and X-ray structure of the protein are almost superimposable.
Figure: NMR Structure of Proteins